单细胞分析流程串讲(Seurat)
本文为单细胞分析系列文章的第三篇。
单细胞转录组测序(scRNA-seq)已成为解析细胞异质性的核心工具,而 Seurat 则是该领域最广泛使用的 R 语言分析包。本文结合 Seurat 官方教程、教材《单细胞组学基础》(樊龙江等)以及笔者在实际项目中的踩坑经验,梳理一份从基本原理到下游分析的完整实战指南。
参考:
一、基本原理
在开始敲代码之前,理解 Seurat 背后的逻辑至关重要。
Seurat 是一个专为单细胞基因表达数据设计的 R 包。它的核心优势在于提供了一个统一的对象结构(Seurat Object),能够存储原始计数矩阵、标准化数据、降维结果、细胞元数据以及分析结果。这使得数据在不同分析步骤间的传递变得非常流畅。
单细胞分析的本质是对 基因 - 细胞矩阵(Gene-Cell Matrix) 的处理。
- 行(Rows):基因(Features),通常经过筛选的高变基因。
- 列(Columns):细胞(Cells),经过质控的高质量细胞。
- 值(Values):UMI 计数或标准化后的表达量。
Seurat 的工作流遵循一个经典范式:
原始数据 -> 质控 (QC) -> 标准化 (Normalization) -> 特征选择 -> 缩放 (Scaling) -> 降维 (PCA) -> 聚类 (Clustering) -> 可视化 (UMAP/tSNE) -> 注释 (Annotation)
生物学上,不同的细胞类型表达不同的基因组合。通过数学方法(如 PCA 和图聚类),我们将表达谱相似的细胞归为一类,这些“计算簇(Cluster)”通常对应着具体的“细胞类型(Cell Type)”。
二、预处理
在正式进入 Seurat 流程前,我们需要明确预处理的目标。这一步决定了分析的上限。低质量细胞会引入噪音,必须剔除。主要关注以下三个指标:
- nFeature_RNA:每个细胞检测到的基因数。
- nCount_RNA:每个细胞检测到的 UMI 总数。
- percent.mt:线粒体基因表达比例。
此外,还需要对样本进行标准化和去批次的操作。这一部分的知识点,可以参考博客往期文章 《RNA-seq数据的归一化》和 《单细胞数据处理中的归一化和去批次效应》 。简单来说:
- 标准化(Normalization):消除测序深度(Sequencing Depth)的影响。例如,细胞 A 测了 1 万条读数,细胞 B 测了 5 万条,直接比较计数是不公平的。
- 批次效应(Batch Effect):不同实验时间、不同操作员或不同芯片产生的技术噪音。如果不去除,聚类结果可能会按“批次”而非“细胞类型”分开。
三、Seurat处理流程
参考: Seurat - Guided Clustering Tutorial
(一)数据读取
通常,10x Genomics 测序数据经过 CellRanger 处理后,会输出包含 barcodes, features, matrix 的文件夹。
1 | library(Seurat) |
经验之谈:如果是多个样本合并,建议先分别创建对象,后续使用
merge或IntegrateData进行整合,不要一开始就合并计数矩阵,以免丢失样本来源信息。
(二)预处理、质控、数据缩放与去批次效应
1. 质控与过滤
1 | # 计算线粒体基因比例 (人类基因以 MT- 开头,小鼠以 Mt- 开头) |
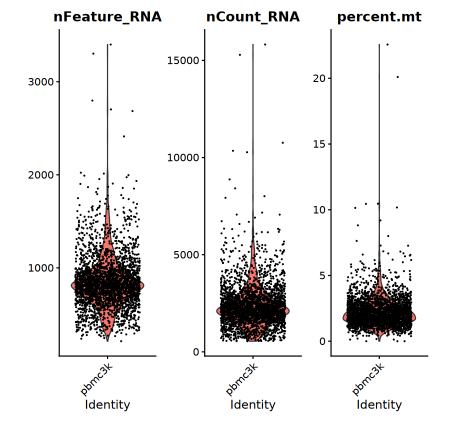
在质控这一步,可以通过调整 subset() 的参数,实现对不同阈值下的样本质控。关于阈值选择,我们可以结合 VlnPlot() 的输出进行调整。
- nFeature_RNA:每个细胞检测到的基因数。
- 过低:可能是空液滴(Empty Droplet)或破碎细胞。
- 过高:可能是双细胞(Doublet)或多细胞粘连。
- nCount_RNA:每个细胞检测到的 UMI 总数。
- 通常与 nFeature 正相关,用于辅助判断细胞捕获效率。
- percent.mt:线粒体基因表达比例。
- 过高:通常意味着细胞膜破损,胞质 mRNA 流失,仅剩下线粒体 mRNA,提示细胞处于凋亡或应激状态。
- 注意:不同组织阈值不同(如肌肉组织本身线粒体含量高,阈值需放宽)。
如果样本是单细胞测序(single cell RNA-seq,scRNA-seq),我们可以不限制 percent.mt 或者使用更为宽松的 percent.mt 阈值(比如说 <5 );对于单细胞核测序(single nucleus RNA-seq,snRNA-seq),由于正常情况下线粒体DNA不太可能出现在细胞核当中,我们可以使用更为严格的 percent.mt 阈值(比如说 <1 )。
2. 标准化与高变基因选择
Seurat 提供了两种主流流程:标准 LogNormalize 和 SCTransform。推荐新手使用 SCTransform,它在标准化和方差稳定化方面表现更佳。
1 | # 方法 A: 标准流程 |

3. 数据缩放 (Scaling)
将基因表达量转换为 Z-score,使所有基因在降维时权重相等。
1 | all.genes <- rownames(x = pbmc) |
4. 去批次效应 (Integration)
如果有多个样本,需进行整合。如果只有单样本,可跳过此步。多样本整合是单细胞分析中最容易出错的环节,务必检查整合后的 UMAP 是否混合了不同样本的细胞。
常用的去批次方法有两种,一种是基于AnchorSet的方法(Seurat自带),另一种是Harmony方法(需要安装第三方库)。目前用Harmony做矫正的研究更多,其使用方法如下:
1 | # 1. merge |
这两种方法的比较,详见 《单细胞数据处理中的归一化和去批次效应》 。
(三)降维、聚类、可视化
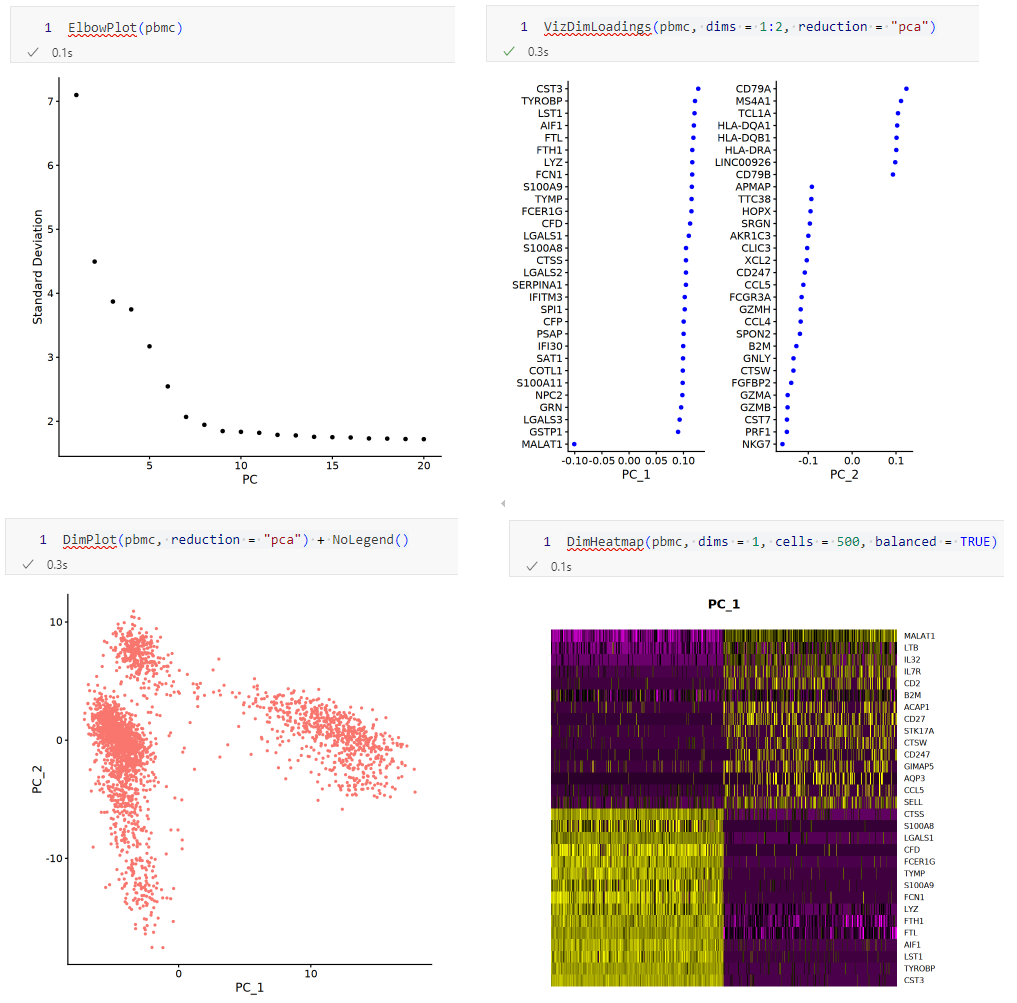
1. 线性降维 (PCA)
1 | pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc)) |

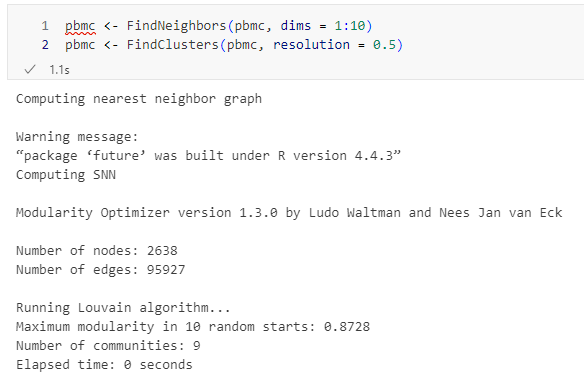
2. 聚类 (Clustering)
基于 K 近邻图(KNN)进行 Louvain 或 Leiden 算法聚类。
1 | pbmc <- FindNeighbors(pbmc, dims = 1:10) # 根据 ElbowPlot 选择 dims |
3. 非线性降维可视化 (UMAP/tSNE)
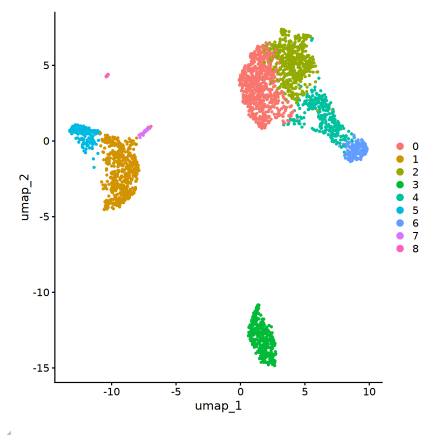
早年间大家用t-SNE的比较多,现在一般用UMAP。根据教材的说法(如下图),UMAP降维的各个类群更为集中,有助于展示不同细胞群体之间的差异。
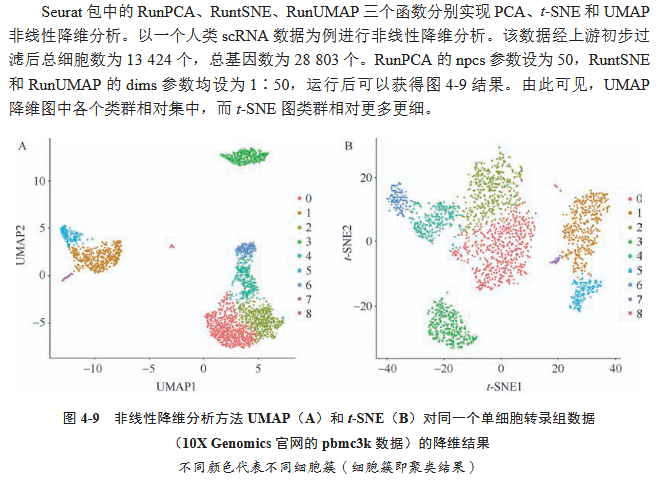
UMAP的代码如下:
1 | pbmc <- RunUMAP(pbmc, dims = 1:10) |
(四)细胞身份注释、Doublet 细胞排除
这一步是单细胞分析的重中之重。经过前一步的处理,我们将细胞分成了许多个类群,但我们并不知道这些类群对应的具体是哪些细胞;而确定这些细胞的身份,我们才能进行后续的生物学功能分析。
1. 寻找标记基因 (Marker Genes)
不同细胞类型,由于需要承担的生物学功能各异,因此会表达不同的基因(例如胚胎期脑组织的Cajal细胞需要发挥结构引导的作用,因此需要表达Reln这种结构蛋白基因;在成熟的兴奋性神经元中,谷氨酸受体相关基因则会高表达)。找到每一群细胞的标记基因,有助于我们确定细胞的身份。
1 | # find markers for every cluster compared to all remaining cells, report only the positive |
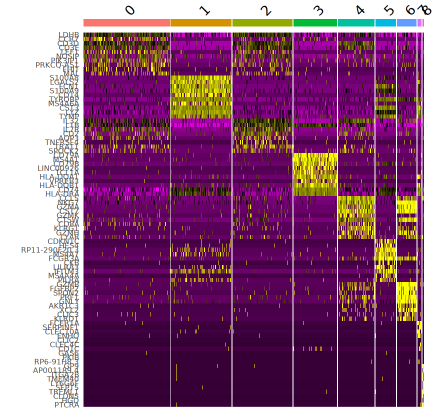
2. 细胞注释 (Annotation)
这是最依赖生物学知识的一步。
- 手动注释:根据
FindAllMarkers结果,结合经典标记基因(如 T 细胞 CD3D, B 细胞 MS4A1, 单核细胞 CD14)进行命名。 - 自动注释:使用
SingleR或scmap包,参考已知数据库(如 Human Primary Cell Atlas)进行预测。
1 | # 示例:重命名细胞群 |
常用自动化注释工具如下:
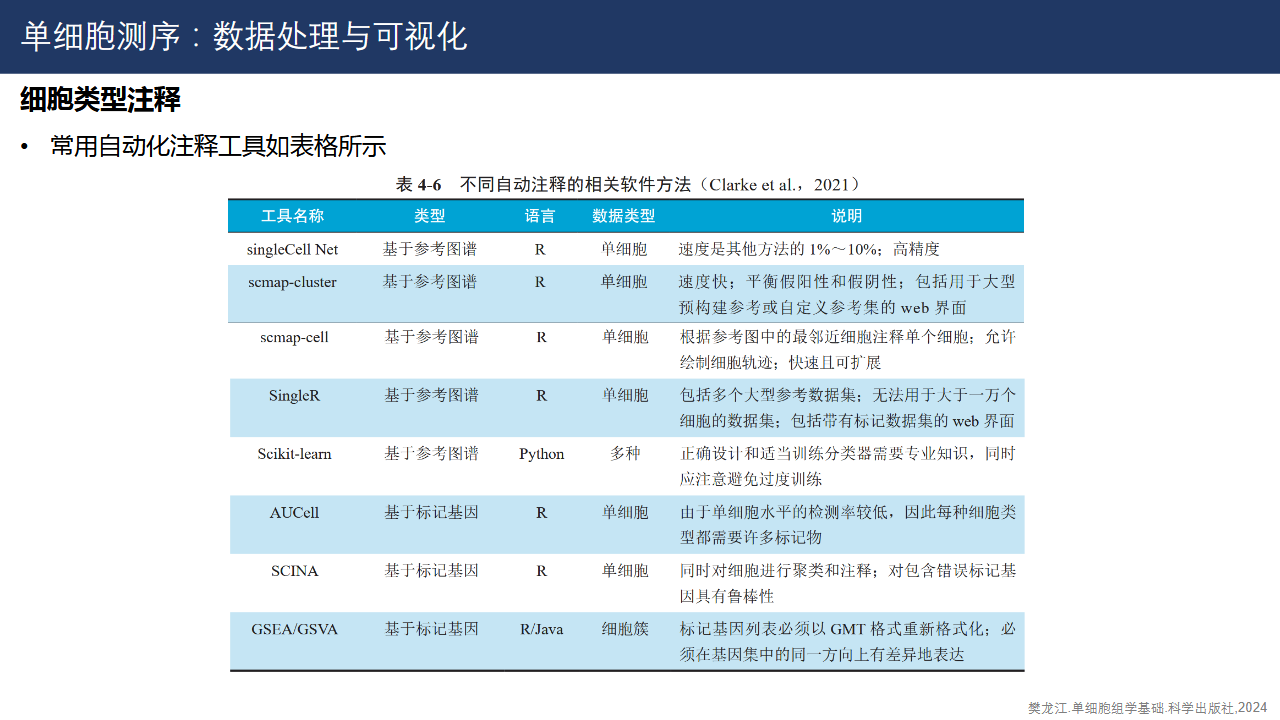
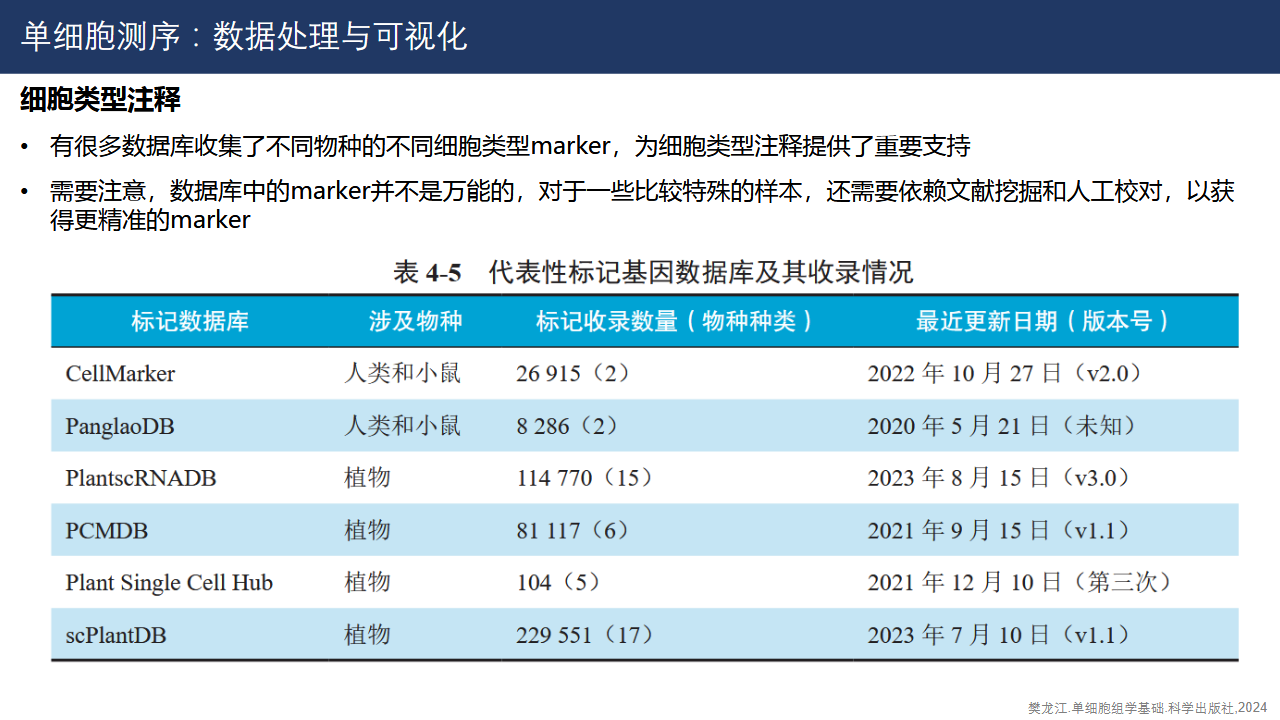
自动化注释对于参考数据库的要求比较高,因此更常用的方法是结合文献挖掘和已知数据库的手动注释。下图展示了常用的单细胞注释的数据库,其中由哈尔滨医科大学整理和发布的CellMarker 是一个相对比较全面的数据库,可以从这里寻找到合适的标记基因。另外,在线富集分析网站Enrichr也提供细胞类型注释的工具,可以作为辅助工具使用。
如何查看特定细胞簇的标记基因呢?常用的统计图包括小提琴图、气泡图、热图和FeaturePlot。在这4种图表中,又以小提琴图和气泡图最为常用。这两种图表一般需要预先知道一系列标记基因,然后通过可视化这些标记基因在不同细胞簇的表达情况,从而推定细胞簇的身份。
例如,现在我们通过检索CellMarker ,知道了一些特定的细胞标记物:
| Marker | Cell type |
|---|---|
| MS4A1 | B cell |
| GNLY | NK cell |
| CD3E | T cell |
| CD14 | Monocyte |
| FCER1A | Dendritic cell |
| FCGR3A | Monocyte/NK cell |
| LYZ | Monocyte |
| PPBP | Platelet |
| CD8A | CD8+ T cell |
我们使用Seurat函数对分群结果进行可视化:
1 | VlnPlot(pbmc, features = c("MS4A1", "GNLY", |
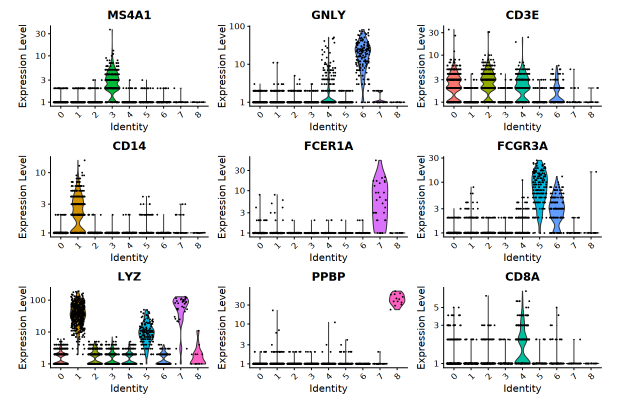
根据biomarker表达情况,我们大致可以做出如下的细胞身份推断。需要注意的是,一些cluster中可能会同时表达多种marker,这种情况可能意味着分辨率太粗(细胞类型没有分开),或者细胞群体本身处于未分化的中间状态,或者某种biomarker确实是多种细胞类型共表达的基因。因此,具体的细胞身份判断还需要依靠生物学知识和实际经验。
| Cluster | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| Putative cell type | T cell | Monocyte | T cell | B cell | CD8+ T cell | Monocyte/NK | NK cell | Dendritic cell | Platelet |
我们也可以使用网站Enrichr辅助判断。例如,前面Cluster5的状态判断存在怀疑,于是我们可以先使用下面的代码提取出cluster5的显著marker:
1 | # 打印前20个marker用于展示 |
cluster5的top20marker如下:
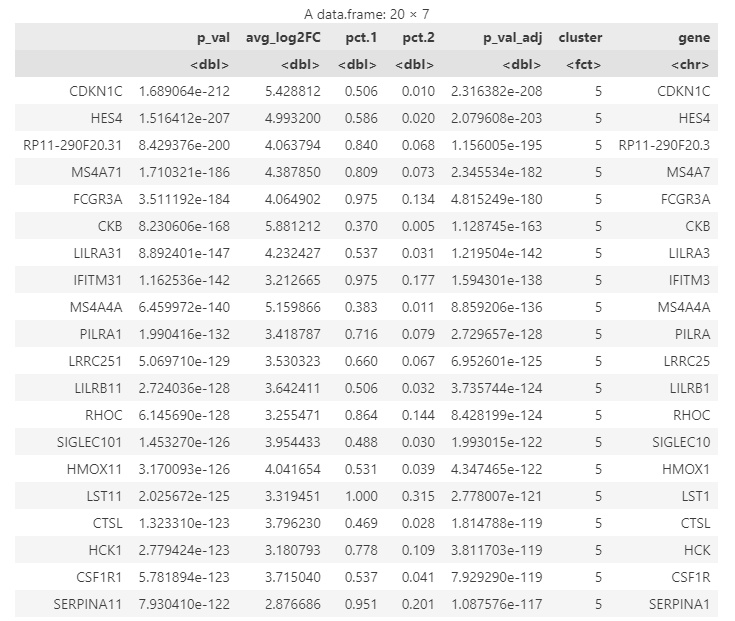
粘贴到Enrichr的输入框:
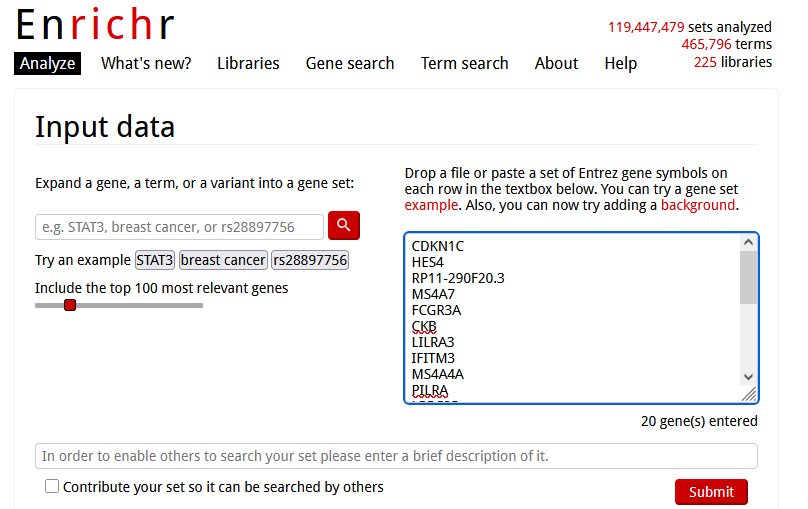
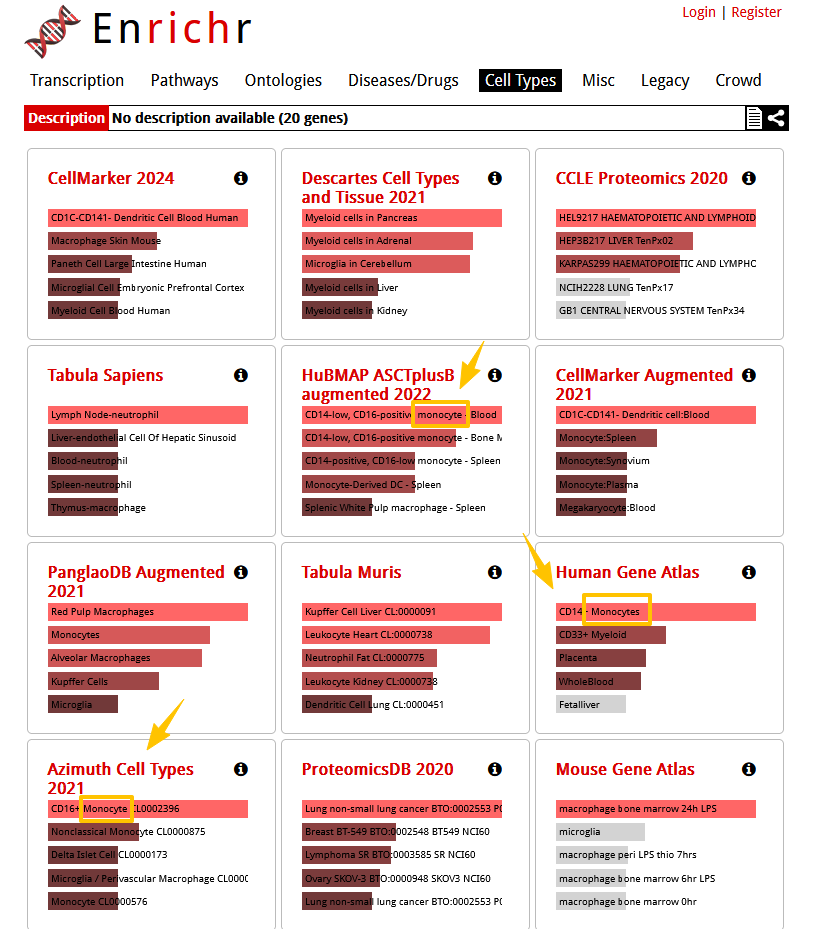
提交查询以后,切换到“Cell Types”标签页,此处就是Enrichr在不同单细胞注释数据集上富集出来的结果。需要注意,这些数据集各自收集的样本来源不同(例如来自小鼠样本),需要根据我们自己数据的实际情况有选择的查看富集结果。如下图,有好几个数据集上都富集出来了monocyte(单核细胞)这一结果,而其他的数据库也给出了和免疫细胞有关的注释结果。因此,Cluster5的细胞大概率就属于monocyte。
3. Doublet 排除
双细胞(Doublet)是指两个细胞被包裹在同一个液滴中。
- 预防:上机时控制细胞浓度。
- 计算排除:使用
DoubletFinder或Scrublet(Python)。通常在聚类前或初步聚类后运行,识别并移除表达谱异常的“杂交”细胞群。
关于 DoubletFinder 的安装和使用,限于篇幅此处不详细探讨,感兴趣的读者朋友可以点击 链接 查看其GitHub代码库主页。
(五)下游分析
完成注释后,真正的生物学挖掘才开始。
- **差异表达分析 (DEG)**:比较不同条件(如 疾病 vs 健康)下同一细胞类型的基因表达差异。
1
2
3# 设置身份为 细胞类型 + 刺激条件
Idents(pbmc) <- "stim"
stim.markers <- FindMarkers(pbmc, ident.1 = "Stim", ident.2 = "Ctrl", subset.ident = "CD4 T") - 细胞通讯分析:使用
CellChat或CellPhoneDB推断细胞间的配体 - 受体互作。 - **拟时序分析 (Trajectory)**:使用
Monocle 3或Slingshot分析细胞分化轨迹。 - 功能富集分析:对差异基因进行 GO/KEGG 富集,解释通路变化。
结语
Seurat 的单细胞分析流程是一个迭代的过程。
- 质控阈值不是一成不变的,需要根据组织类型调整。
- 聚类分辨率(Resolution)需要多次尝试,以找到生物学意义最明确的分群。
- 细胞注释往往需要结合文献和多个标记基因综合判断,切勿仅凭单一基因定论。