单细胞数据处理中的归一化和去批次效应
如题。在实验数据处理中,我们经常会碰到归一化和去批次的问题。对于RNA-seq数据的归一化,常用的方法有RPKM矫正,TPM矫正,Quantile Normalization等(参见博客往期文章《RNA-seq的数据归一化》 ),而单细胞测序数据,作为一类更加复杂的组学数据,其归一化和去批次的方法也更多更复杂。以下是结合近期实践的一些经验分享,写得不好,请多见谅。
一、归一化
参考:
单细胞转录组的归一化方法大概可以分成两大类。一类是基于“size factors”的方法,例如LogNormalize,即直接除以总数的log标准化;另一类是基于概率分布的方法,根据每个基因的表达量分布拟合特定的参数,然后对每个基因标准化,例如SCTransform。
这两大类标准化方法侧重的维度不同,size factors 侧重细胞维度;概率分布侧重基因维度。
(一) LogNormalize in Seurat
其使用方法如下:
1 | library(dplyr) |
归一化之后的表达矩阵数据的存储槽位为 SeuratObject@assays$RNA@layers$data 。
函数实现见Github存储库 , API说明见Seurat文档。下面是核心逻辑:
1 | # File seurat/R/preprocessing5.R, Line 188-220. |
LogNormalize使用的归一化公式为
$$
\text{xnorm} = \log_1p\left(\frac{x}{\sum x} \times \text{scale.factor}\right)
$$
x/sum(x):将当前单元(如细胞)的每个基因的UMI计数转换为比例。scale.factor:将总表达量缩放至目标值(如1e4)。log1p:对结果取自然对数(log1p(x) = log(x + 1)),避免零值问题。
LogNormalize是Seurat中默认的归一化方法,其做了两件事:①消除测序深度差异(如不同细胞的UMI总量差异);②压缩数据动态范围,便于后续分析(如聚类、降维)。
核心算法原理非常简单,计算速度也是最快的。但缺点是只矫正了测序深度,并没有矫正异常基因。
(二) SCTransform in Seurat
一个示例如下:
1 | # store mitochondrial percentage in object meta data |
这是一个用方差稳定变换对单细胞UMI count 数据标准化的方法,方差稳定变换是基于负二项回归。这个函数在对数据进行均一化的同时还可以去除线粒体红细胞等混杂因素的影响。
Sctransform先拟合每个基因在不同细胞的reads数和测序深度的关系,然后矫正拟合结果,计算测序深度相关的reads数期望值和对应的方差,利用期望值和方差计算皮尔森残差,最后返回矫正后的reads数。
SCTransform的优点:
- 不根据size factor,也就是总的UMI数对每个细胞标准化;
- 标准化方法建立在单细胞数据已知的关系之上:平均表达量和方差的关系。
- 没有用log转化,z-scoring标准化(严格来说,sctransform还是有涉及的。计算皮尔森残差的公式很像z-score)
- 综合来说,sctransform的标准化结果可以消除测序深度的影响。
SCTransform标准化的过程大概分为三步(如下图,另外参考文章):
- 负二项分布回归(广义线性模型)拟合每个基因的表达量和测序深度的关系;
- Ksmooth矫正拟合结果;
- 计算皮尔森残差,返回矫正reads数。

另外 ,有人指出SCTransform只需要运行一遍即可,在整合去批次以后的样本上不应该再次运行SCTransform( "Do not run a second round of SCTransform on the integrated assay" ,参考 issues1836 ),这是因为SCT 归一化是针对每个样本单独进行的,因此可能会引入批次效应( "SCT normalization was done separately for each sample, which is likely to introduce batch effects down the line" , 参考 issues2023 )。
另外,在这个方法中有一个可选参数 vars.to.regress ,在上述Seurat示例中使用的是 vars.to.regress = "percent.mt" ,它表示在建立负二项分布模型时将线粒体基因占比(percent.mt) 也作为一个协变量添加到模型当中,从而在归一化时移除线粒体基因占比造成的差异。这个参数可以换成其他指标,例如某个管家基因的表达量,或者细胞数量等等。如果不需要考虑这些差异,仅仅需要矫正测序深度的影响,则可以留空。
另外是AI大模型总结的一张表格,对比了两个方法的优缺点:(powered by DeepSeek)
SCTransform vs LogNormalize:
| 特征 | LogNormalize | SCTransform |
|---|---|---|
| 核心原理 | 全局缩放和对数变换 | 正则化负二项回归模型 |
| 关键假设 | 所有细胞的初始RNA总量相近 | 基因表达计数遵循负二项分布,且受测序深度影响 |
| PCA输入 | 对数变换后的数据(data槽) | 皮尔逊残差(scale.data槽) |
| 处理测序深度 | 隐式处理(作为分母),假设其为唯一技术因素 | 显式处理(作为模型协变量),更精确地分离技术与生物变异 |
| 主要优势 | 概念简单,计算速度快 | 统计学基础扎实,方差稳定效果好,能更有效去除技术噪音 |
| 潜在不足 | 核心假设常被违背,可能扭曲生物学差异 | 模型相对复杂,计算成本较高,在特定模拟数据中可能存在偏好 |
总之,推荐的归一化方法是SCTransform。如果数据量比较大,需要节省时间,或者数据中没有太多高表达的异常基因,则可以使用 LogNormalize。
二、批次效应移除
如果我们有多个样本,需要合并在一起,那么样本之间如何保证可比?不同样本(即使来自相同的测序平台),其样本状态、测序深度、技术误差等等因素叠加在一起,也会导致很强的批次效应。下面介绍几种R语音中常用的批次效应移除工具。
(一) 基于锚点的去批次方法(Integrate AnchorSet by IntegrateData in Seurat)
参考: Stuart T, Butler A, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888-1902 doi:10.1016/j.cell.2019.05.031
某种意义上说,IntegrateData 这一类方法与前面介绍的那些并不相同,它为多组样本的整合任务而生,主要目的是去除样本之间的批次效应。
通过识别跨数据集的单细胞之间的成对对应关系(称为“锚点”),我们可以将数据集转换到一个共享的空间,这使得能够在组织或生物体尺度构建协调一致的图谱,并且能够有效地将离散或连续的数据从参考数据集转移到查询数据集上。
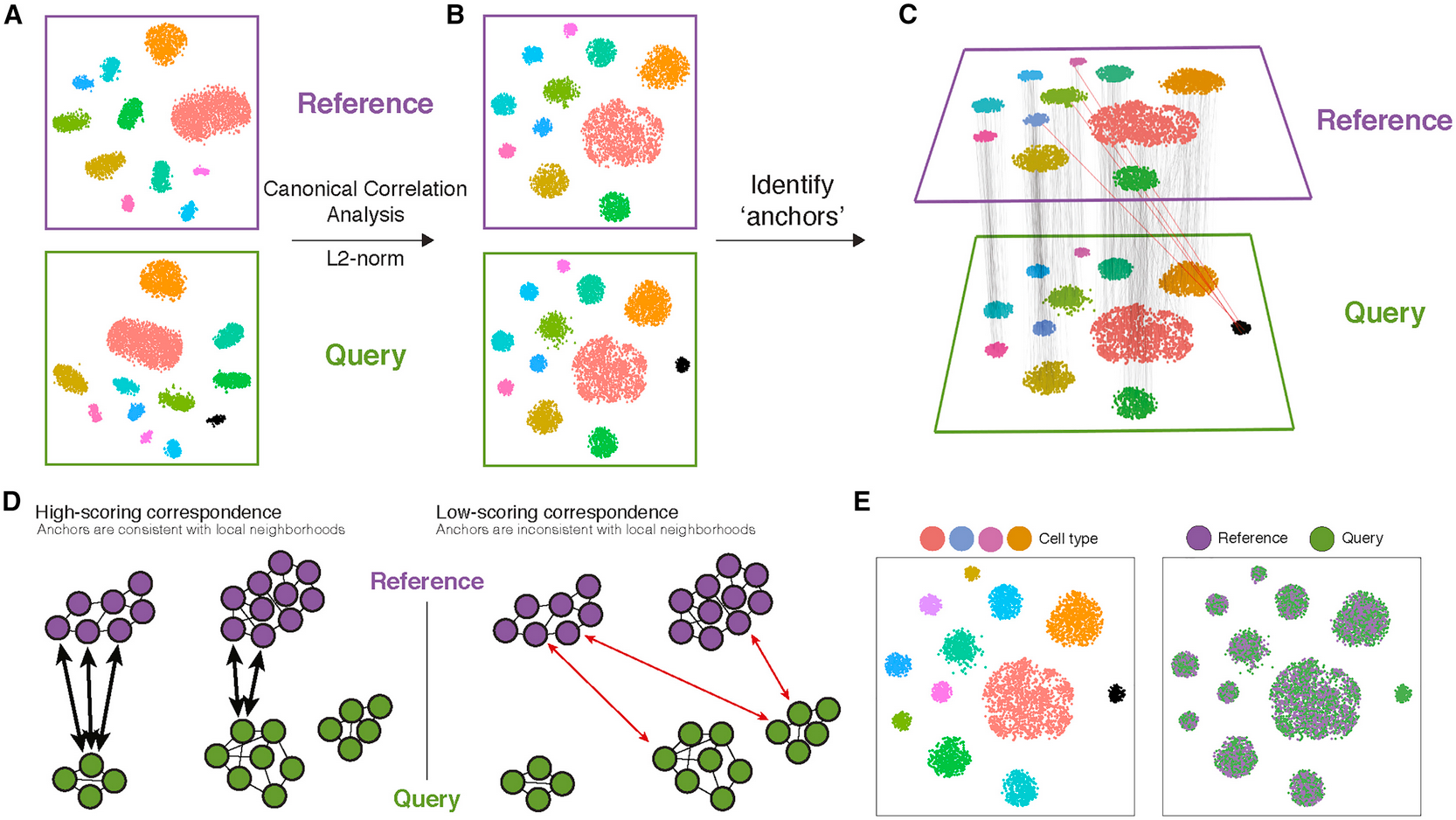
具体来说,基于锚点的去批次方法包含如下几个步骤(如上图)
- 首先,我们的输入包括两个数据集(参考数据集和查询数据集),每个数据集都来源于单独的单细胞实验。这两个数据集共享来自相似生物状态的细胞,但查询数据集还另外包含一个独特的细胞群体(图A中以黑色标注的细胞簇)。
- 对这些细胞进行经典的相关性分析,随后进行 L2 归一化,将数据集投影到由跨数据集的共享相关结构定义的子空间中(B图)。
- 在共享空间中,我们在参考细胞和查询细胞之间识别成对的 MNN(最近邻对),它们代表跨数据集处于相同生物状态的细胞(灰色线),并充当引导数据集集成的锚点。原则上,独特群体中的细胞不应参与锚点,但在实践中,我们观察到频率较低的“错误”锚点(红色线)。(C图)
- 对于每对锚点,我们根据各个数据集的邻域结构中锚点的一致性来分配一个分数。我们利用锚点及其分数为每个查询细胞计算“校正”向量,从而转换其表达,使其能够作为集成参考的一部分进行联合分析。(D-E图)
要使用这套方法进行批次效应移除,我们的步骤如下:
1 | # 0. 合并多个样本 |
(二) RunHarmony in Seurat and Harmony
参考:
- Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). https://doi.org/10.1038/s41592-019-0619-0
- Harmony Integration
- https://cran.r-project.org/web/packages/harmony/index.html
- https://cran.r-project.org/web/packages/harmony/harmony.pdf
- https://cran.r-project.org/web/packages/harmony/vignettes/Seurat.html
Harmony是2019年Korsunsky等人提出的一种去批次的方法,其计算思路大致为「低维嵌入→软聚类分组→计算聚类质心→计算校正因子→细胞特异性线性因子校正→迭代直到收敛」。下图是论文中给出的算法大纲。
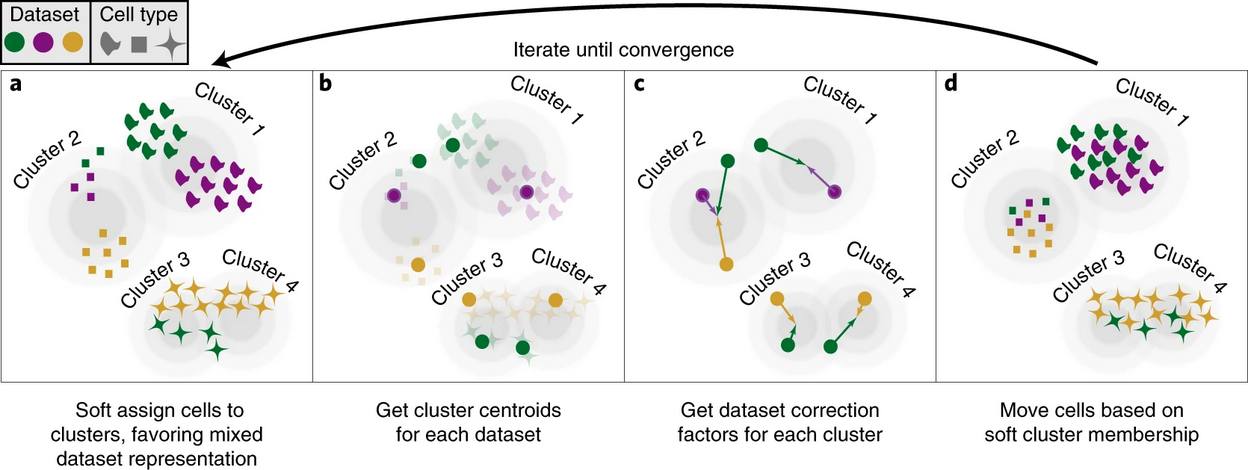
这个方法的计算细节详见论文,此处不做展开。Harmony方法的优点在于速度很快,不过相比于前述Seurat中基于锚点的方法,Harmony方法只能返回去批次以后的细胞坐标,而无法返回基因表达矩阵,因此如果对于跨数据集的表达量有比较的需求,则不建议使用这种方法。
使用方法如下:
1 | # 1. merge |
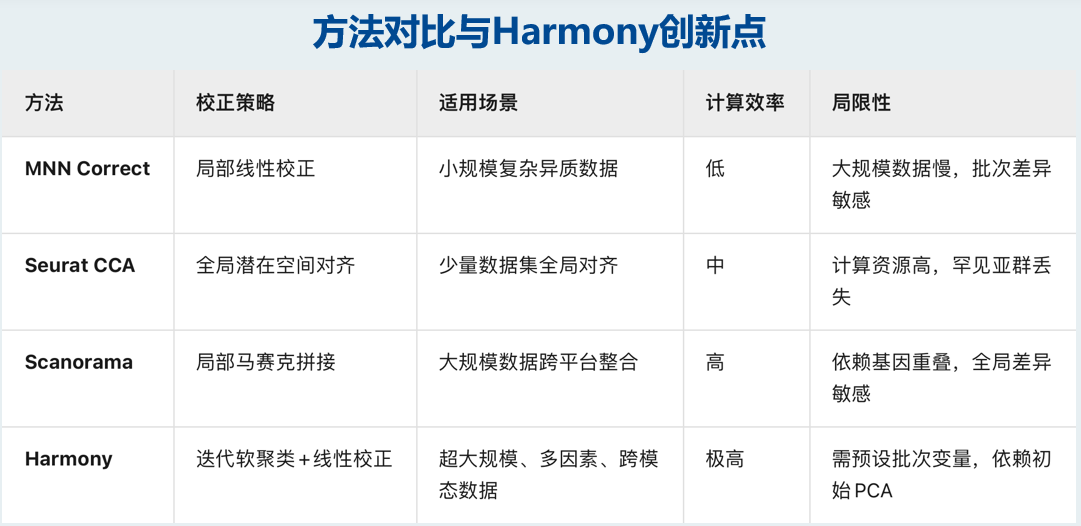
再附一张图,关于几种方法的比较。
(三) 其他方法
参考:
- Shi, Q., Chen, Y., Li, Y. et al. Cross-tissue multicellular coordination and its rewiring in cancer. Nature (2025). https://doi.org/10.1038/s41586-025-09053-4 ; https://www.nature.com/articles/s41586-025-09053-4#citeas
- 新概念新方法新发现—张泽民课题组揭示跨组织多细胞协同模式及其在肿瘤中的重塑
在张泽民老师刚刚发表的论文中,研究团队整合分析了多个单细胞数据,揭示了肿瘤发生过程中多细胞之间的协同模式。在论文methods部分,研究团队测试了多种数据整合和去批次方法,并对这些方法的性能进行了一些的说明。
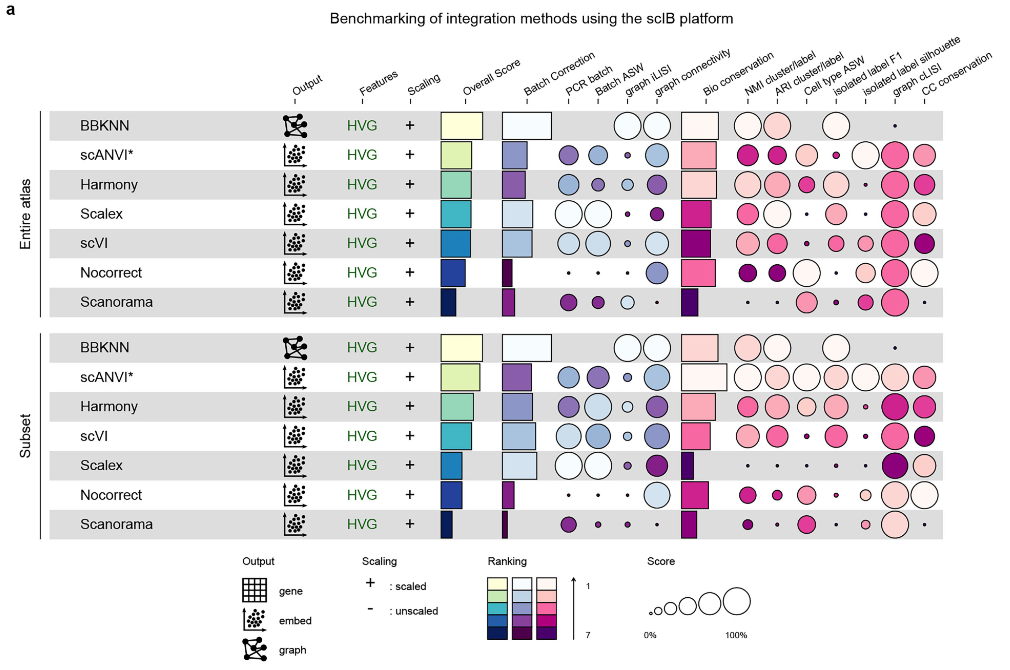
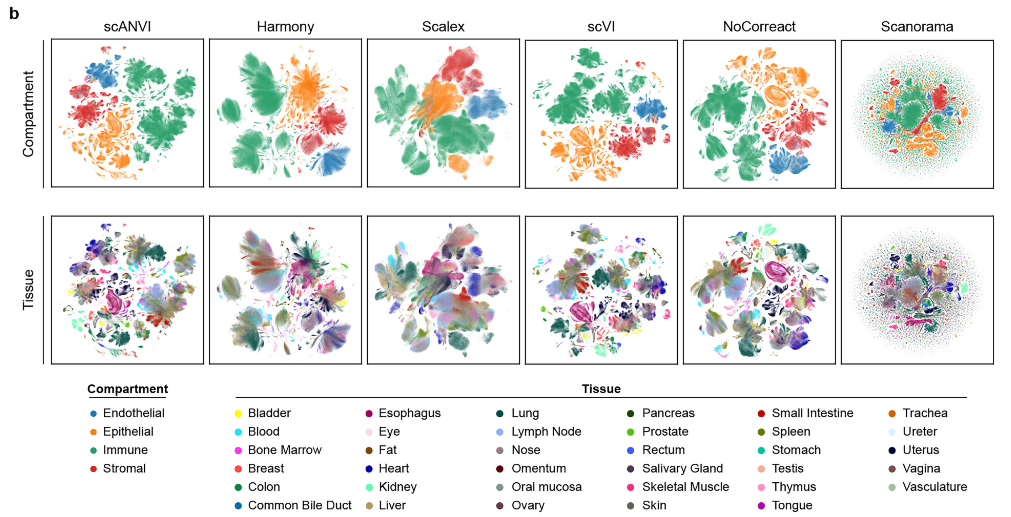
如上图。作者使用scIB 17 这一方法,评估了几种广泛使用的基于 Python 的工具:BBKNN 18 、Harmony 51 、Scanorama 52 以及基于深度学习的 scVI 53 、scANVI 54 和 SCALEX 55 。最终,BBKNN 表现出色,被用于整合跨组织的数据集。
从图中可以看出,除了BBKNN以外,其实scANVI、Harmony和scVI也有比较好的效果。另外,这些工具是基于python的,一般会与scanpy的流程合并起来使用;如果使用Seurat这套流程做分析,或许其中的一些方法(例如张泽民这篇文章中推荐的BBKNN)可能就不适用了。
三、一些注意事项
需要注意,归一化方法,尤其是SCTransform方法,是针对单个样本进行的。即使数据已经进行了合并,归一化也是只在单个样本上进行。
因此,在Github上,有人指出,针对多个样本的归一化处理,SCTransform只需要运行一遍即可,在整合去批次以后的样本上不应该再次运行SCTransform( "Do not run a second round of SCTransform on the integrated assay" ,参考 issues1836 ),否则,可能会引入批次效应( "SCT normalization was done separately for each sample, which is likely to introduce batch effects down the line" , 参考 issues2023 )。
以上。