文献阅读笔记:基于生物学信息的神经网络
Selby, D.A., Sprang, M., Ewald, J. et al. Beyond the black box with biologically informed neural networks. Nat Rev Genet (2025). https://doi.org/10.1038/s41576-025-00826-1
Beyond the black box with biologically informed neural networks
(打破黑箱:基于生物学信息的神经网络)
【按:从去年到现在,断断续续看到了许多篇关于神经网络在生物信息学中应用的论文。在这方面,常常面临一个问题:”模型预测的精确性”与”模型可解释性”的权衡。一种解决思路是先训练模型,然后用机器学习可解释方法去对黑箱模型进行解读;另一种解决思路就是引入生物学信息,构建所谓的Biologically informed deep neural network(如下图,来自P-Net模型的论文)。本文发表于本月初的nature reviews genetics期刊上,在文章中作者总结归纳了后一类模型,并在参考文献部分列出了几篇有代表性的论文。】
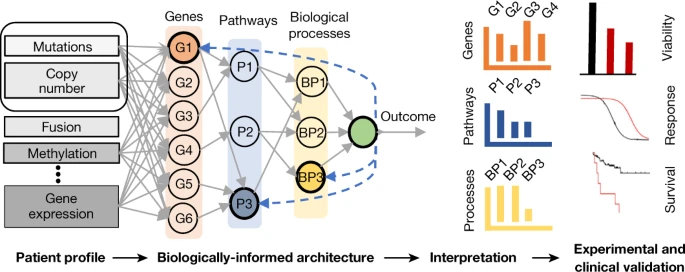
Machine learning models for multi-omics data often trade off predictive accuracy against biological interpretability. An emerging class of deep learning architectures structurally encode biological knowledge to improve both prediction and explainability. Opportunities and challenges remain for broader adoption.
多组学数据的机器学习模型通常在预测准确性和生物可解释性之间进行权衡。一类新兴的深度学习架构通过结构化地编码生物学知识来提高预测和可解释性。但要更广泛地采用,仍存在机会和挑战。
Machine learning applied to data from high-throughput technologies has transformed biological research, facilitating integration of diverse omics datasets into multi-layered models of cellular systems. However, the predictive power of traditional ‘black box’ machine learning algorithms — and their ability to model complex nonlinear relationships — often comes at the expense of biological interpretability. Biologically informed neural networks (BINNs) offer a promising solution, combining predictive accuracy with explainability by incorporating decades of accumulated prior biological knowledge. This emerging paradigm is particularly well-suited for understanding model predictions based on complex, high-dimensional multimodal datasets found in multi-omics integration.
应用于高通量技术数据的机器学习已经改变了生物学研究,促进了将各种组学数据集整合到多层次的细胞系统模型中。然而,传统“黑箱”机器学习算法的预测能力——及其建模复杂非线性关系的能力——通常以生物可解释性为代价。结合了数十年积累的先前生物知识的生物信息神经网络(BINNs)提供了一个有前景的解决方案,通过这种方法可以在保证预测准确性的同时实现可解释性。这一新兴范式特别适用于理解基于在多组学整合中发现的复杂、高维多模态数据集的模型预测。
From black boxes to visible neural networks
(从黑盒子到可见的神经网络)
BINNs are artificial neural networks whose architecture is explicitly constrained by biological pathway ontologies (Fig. 1). Unlike conventional fully connected deep learning models, which rely on arbitrarily chosen numbers of hidden nodes and layers, BINNs are designed using known pathway hierarchies from databases such as Reactome, Gene Ontology or KEGG1. Each node in the network represents a real-world biological entity — such as a gene, pathway or biological process — and edges reflect known relationships between these entities. For instance, an input node representing gene expression levels is only connected to a hidden pathway node if the gene is a known member of that pathway. This structure, which contrasts with the opacity of traditional black box models, has led to the term visible neural networks2,3, or transparent neural networks4.
BINNs 是人工神经网络,其架构明确受到生物路径本体论的约束(图 1)。与依赖于任意选择的隐藏节点和层数的传统全连接深度学习模型不同,BINNs 使用来自数据库(如 Reactome、Gene Ontology 或 KEGG)中已知的路径层次结构进行设计 1 。网络中的每个节点代表一个现实世界中的生物实体——例如基因、路径或生物过程——边反映了这些实体之间的已知关系。例如,表示基因表达水平的输入节点仅在基因是该路径已知成员的情况下才连接到隐藏路径节点。这种结构与传统黑盒模型的不透明性形成对比,导致了可见神经网络 2,3 或透明神经网络 4 的术语。
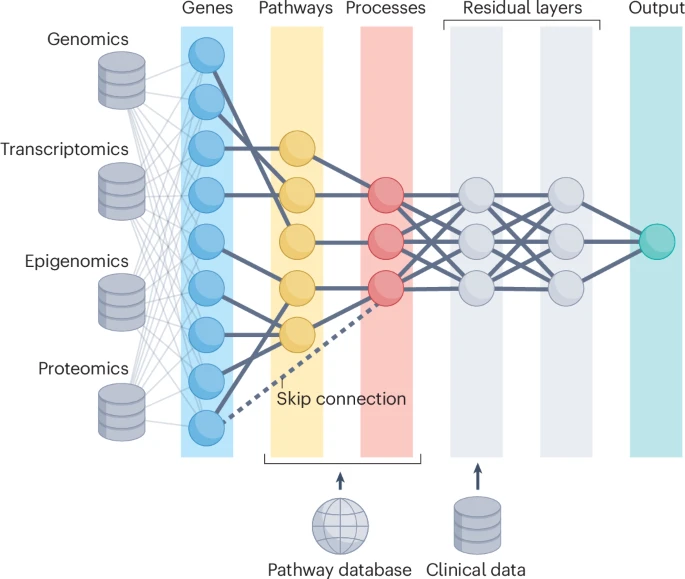
Figure1: Multiple omics are used as input and their respective features are linked to genes via known mappings. The genes are, in turn, connected to a hierarchy of biological ontology from a database (for example, genes, pathways and higher-order processes). To accommodate uneven hierarchies, skip connections (dotted line) or dummy nodes can be used. Fully connected residual nodes may capture interactions not included in the structural ontology used to build the architecture, which otherwise embeds a strong inductive bias in the predictions. Clinical measurements, or other data which cannot be linked directly to the pathway ontology, can be included via late fusion into the neural network or via dummy pathways.
图1: 多种组学被用作输入,它们各自的特征通过已知的映射链接到基因。这些基因又与来自数据库的生物本体论层次结构中的项相关联(例如,基因、通路和更高级别的过程)。为了适应不均匀的层次结构,可以使用跳过连接(虚线)或虚拟节点。完全连接的残差节点可能捕捉到未包含在用于构建架构的结构本体论中的交互作用,否则这些预测会嵌入强烈的归纳偏差。临床测量或其他无法直接链接到通路本体论的数据可以通过晚期融合纳入神经网络,或者通过虚拟通路实现。
The biologically informed architecture of BINNs tackles several challenges simultaneously. First, the incorporation of well-curated biological knowledge reduces the number of model parameters, thereby decreasing the amount of training data required. Second, the structure of the model is intuitive for biomedical researchers, even those with limited machine learning expertise. Third, the reduced dependence on training data and inductive bias mitigates overfitting and increases generalizability. Finally, by emulating cellular and genetic regulation, BINNs bridge the gap between data-driven models and mechanistic biological understanding.
具有生物学信息的 BINNs 架构同时解决了多个挑战。首先,整合经过良好整理的生物学知识减少了模型参数的数量,从而减少了所需的训练数据量。其次,即使对于有限机器学习经验的生物医学研究人员来说,该模型的结构也是直观易懂的。第三,减少对训练数据和归纳偏置的依赖减轻了过拟合现象并提高了泛化能力。最后,通过模拟细胞和基因调控,BINNs 弥合了数据驱动模型与机制性生物学理解之间的差距。
Applications and success stories of BINNs
(BINNs 的应用和成功案例)
Since their introduction around 2018, BINNs have been widely applied in biomedicine, with notable successes in oncology, drug response prediction and survival analysis1,4. For example, models such as P-Net5 have demonstrated efficacy in aligning molecular features with therapeutic outcomes. Other extensions have integrated genomic data with chemical structure data to predict therapeutic efficacy3 or combined multi-omics and clinical data to predict patient survival in precision medicine6. BINNs are not limited to supervised learning tasks; biologically informed variational autoencoders — a form of unsupervised learning model — have also been used to analyse cellular processes and aid drug development7.
自 2018 年左右引入以来,BINNs 已在生物医学领域得到广泛应用,在肿瘤学、药物反应预测和生存分析方面取得了显著成功 1,4 。例如,P-Net 5 等模型已证明在将分子特征与治疗结果对齐方面具有有效性。其他扩展已将基因组数据与化学结构数据相结合以预测治疗效果 3 ,或将多组学和临床数据结合起来以预测精准医疗中的患者生存率 6 。BINNs 不仅限于监督学习任务;生物信息变分自动编码器——一种无监督学习模型——也被用于分析细胞过程并辅助药物开发 7 。
Recent applications have extended BINNs to single-cell sequencing, uncovering cellular heterogeneity and regulatory networks. Although early works already used multi-omics data, the integration of multiple modalities has increased with time. These models have also been used to uncover novel pathway interactions, demonstrating their potential as discovery agents8.
近期的应用已经将 BINNs 扩展到了单细胞测序,揭示了细胞异质性和调控网络。尽管早期的工作已经使用了多组学数据,但随着时间的推移,多种模态的整合越来越多。这些模型还被用于揭示新的通路相互作用,展示了它们作为发现工具的潜力 8 。
Why BINNs excel in multi-omics integration
(为什么生物信息神经网络(BINNs)在多组学整合方面表现出色)
Multi-omics datasets are inherently high-dimensional, heterogeneous and often limited in sample size relative to the number of features. BINNs leverage biological priors to reduce model complexity by constraining the hypothesis space early in the analysis pipeline, which — ideally — improves generalizability and predictive performance and makes them particularly effective in these scenarios.
多组学数据集本质上是高维的、异构的,并且相对于特征数量而言样本量通常较小。BINNs 利用生物学先验知识,在分析流程的早期阶段限制假设空间以降低模型复杂性,这在理想情况下可以提高泛化能力和预测性能,并使它们在这类场景中特别有效。
Comparative studies reveal that BINNs perform comparably to, or better than, fully connected neural networks on various predictive tasks. For example, BINNs seem to excel in scenarios with small, high-dimensional datasets, which are suboptimal for dense neural networks but typical of omics studies. They also outperform traditional machine learning models in capturing non-linear, hierarchical relationships inherent to biological systems (Fig. 1), enabling meaningful insights beyond prediction, such as discovery of novel biomarkers5,8. Multiple omics fit in this hierarchy, because nodes in BINNs can represent any biological entity, for example, genes, metabolites or protein complexes. In genomic assays, such as mutation or copy number variation measurements, the features are mapped to the gene that contains the aberration. In transcriptomics and proteomics, multiple transcripts or proteins can be mapped to one gene. In metabolomics, a metabolite may be mapped to genes encoding enzymes that use or produce the respective molecule. To accommodate multiple inputs, a common entity is chosen (that is, genes) or specialized input layers can be crafted. As BINNs are special cases of multimodal deep learning, different data fusion strategies can be explored9.
比较研究显示,BINNs 在各种预测任务中表现与全连接神经网络相当或更优。例如,BINNs 在小而高维的数据集上似乎表现出色,这些数据集对密集型神经网络来说效果不佳,但却是基因组学研究中的典型情况。它们在捕捉生物系统固有的非线性、层次关系方面也优于传统机器学习模型(图 1),从而能够在预测之外获得有意义的见解,例如发现新的生物标志物 5,8 。多个组学数据可以纳入这一层次结构,因为 BINNs 中的节点可以代表任何生物实体,例如基因、代谢物或蛋白质复合物。在基因组测定中,如突变或拷贝数变异测量,特征被映射到包含异常的基因。在转录组学和蛋白质组学中,多个转录本或蛋白质可以映射到一个基因。在代谢组学中,代谢物可以映射到编码使用或产生相应分子的酶的基因。为了容纳多个输入,可以选择一个共同的实体(即基因)或设计专门的输入层。 由于生物信息神经网络(BINNs)是多模态深度学习的特例,可以探索不同的数据融合策略 9 。
Moreover, BINNs integrate predictive and explanatory tasks seamlessly. Traditional machine learning models, including dense neural networks, often fail to provide biologically meaningful insights owing to the inexplicability of their internal nodes. Post-hoc, model-agnostic interpretability methods offer input-level explanations but are prone to instability and can fail to reflect highly non-linear relationships10, such as between genes and processes. By contrast, BINNs enforce interpretability as an intrinsic property, allowing predictions to be directly linked to specific genes or pathways. This ante-hoc approach enhances robustness by incorporating known biological constraints, making BINNs ideal for tasks that require both prediction and inference, such as biomarker discovery and drug target validation.
此外,BINNs(生物信息神经网络)能够无缝集成预测和解释任务。传统的机器学习模型,包括密集型神经网络,由于内部节点的不可解释性,往往无法提供具有生物学意义的见解。后验的、与模型无关的解释方法虽然可以提供输入级别的解释,但容易不稳定,并且可能无法反映高度非线性的关系,比如基因与过程之间的关系。相比之下,BINNs 将可解释性作为其内在属性,使得预测可以直接关联到特定的基因或通路。这种方法通过整合已知的生物约束,在先验阶段增强了鲁棒性,使 BINNs 成为需要同时进行预测和推断的任务的理想选择,例如生物标志物发现和药物靶点验证。
Advancing BINNs for biomedical discovery
(推进 BINNs 在生物医学发现中的应用)
Despite their promise, BINNs face several challenges. Most studies evaluate BINNs within narrow datasets and tasks, limiting insights into their generalizability across domains and conditions. The reasons for their apparently superior performance — whether due to biological inductive bias, multi-omics data fusion strategies or the introduced sparsity — remain unclear. Additionally, the lack of standardized benchmarks and tools hampers accessibility and reproducibility.
尽管前景广阔,BINNs 仍面临若干挑战。大多数研究在狭窄的数据集和任务中评估 BINNs,限制了对其跨领域和条件普适性的见解。它们表现优异的原因——无论是由于生物学归纳偏置、多组学数据融合策略还是引入的稀疏性——仍然不清楚。此外,缺乏标准化基准和工具阻碍了可访问性和可重复性。
To fully realize their potential, future research should focus on developing robust frameworks for BINN construction and evaluation. Expanding the use of flexible architectures capable of handling various kinds of biological knowledge, incorporating advanced multimodal fusion strategies and systematically exploring the impact of different ontologies will be essential. Furthermore, leveraging BINNs for hypothesis generation, such as predicting novel pathway relationships, represents an exciting research opportunity.
为了充分发挥其潜力,未来的研究应侧重于开发稳健的 BINN 构建和评估框架。扩展使用能够处理各种生物知识的灵活架构,结合先进的多模态融合策略,并系统地探索不同本体论的影响将是至关重要的。此外,利用 BINN 进行假设生成,例如预测新的通路关系,代表了一个令人兴奋的研究机会。
BINNs may represent a transformative approach in computational biology, uniting predictive accuracy with biological interpretability. By embedding domain knowledge gathered over decades of genetic research, these architectures provide more transparent, data-driven biomedical models that reduce computational costs and enable built-in interpretability. However, to fully harness their potential, the field must address key challenges:
BINNs 可能代表了一种在计算生物学中的变革性方法,它将预测准确性与生物学可解释性相结合。通过嵌入数十年遗传研究积累的领域知识,这些架构提供了更透明的数据驱动生物医学模型,降低了计算成本,并实现了内置可解释性。然而,为了充分发挥它们的潜力,该领域必须应对关键挑战:
- Standardization: develop common benchmarks and tools to improve accessibility, reproducibility and study comparability.(标准化:制定共同的基准和工具以提高可访问性、重现性和研究的可比性。)
- Rigorous evaluation: conduct more comprehensive evaluations and ablation studies to understand the mechanisms behind the performance of BINNs and their generalizability relative to alternative approaches, such as graph neural networks and classic machine learning.(严格评估:进行更全面的评估和消融研究,以理解 BINNs 性能背后的工作机制及其相对于替代方法(如图神经网络和经典机器学习)的泛化能力。)
- Flexible architectures: explore architectures that can incorporate diverse biological knowledge and advanced data fusion strategies.(灵活的架构:探索能够整合各种生物学知识和先进数据融合策略的架构。)
- Hypothesis generation: combining modern neural architecture search methods with BINNs could unlock discovery of novel pathway interactions and regulatory mechanisms.(假设生成:结合现代神经结构搜索方法与 BINNs 可能解锁对新型路径交互和调控机制的发现。)
- Focus on the core: systematically investigate the choice of knowledge databases and the hierarchy level to build BINNs in close relation to their application.(关注核心:系统地研究知识数据库的选择和层次级别,以构建与应用密切相关的生物信息神经网络(BINNs)。)
Overcoming these hurdles may unlock the full potential of multi-omics and BINNs, paving the way for more explainable, data-driven discoveries in genomics, drug development and precision medicine.
克服这些障碍可能会充分发挥多组学和生物信息神经网络的全部潜力,从而为基因组学、药物开发和精准医学中的更多可解释的数据驱动发现铺平道路。
References 参考文献
- Wysocka, M., Wysocki, O., Zufferey, M., Landers, D. & Freitas, A. A systematic review of biologically-informed deep learning models for cancer: fundamental trends for encoding and interpreting oncology data. BMC Bioinformatics 24, 198 (2023).
- van Hilten, A. et al. Phenotype prediction using biologically interpretable neural networks on multi-cohort multi-omics data. npj Syst. Biol. Appl. 10, 81 (2024).
- Kuenzi, B. M. et al. Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell 38, 672–684 (2020).
- Novakovsky, G., Dexter, N., Libbrecht, M. W., Wasserman, W. W. & Mostafavi, S. Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat. Rev. Genet. 24, 125–137 (2023).
- Elmarakeby, H. A. et al. Biologically informed deep neural network for prostate cancer discovery. Nature 598, 348–352 (2021).
- Hao, J., Kim, Y., Mallavarapu, T., Oh, J. H. & Kang, M. Interpretable deep neural network for cancer survival analysis by integrating genomic and clinical data. BMC Med. Genomics 12 (Suppl. 10), 189 (2019).
- Seninge, L., Anastropoulos, I., Ding, H. & Stuart, J. VEGA is an interpretable generative model for inferring biological network activity in single-cell transcriptomics. Nat. Commun. 12, 5684 (2021).
- Hou, Z., Leng, J., Yu, J., Xia, Z. & Wu, L. Y. PathExpSurv: pathway expansion for explainable survival analysis and disease gene discovery. BMC Bioinformatics 24, 434 (2023).
- Nguyen, T. et al. Optimal fusion of genotype and drug embeddings in predicting cancer drug response. Brief. Bioinform. 25, bbae227 (2024).
- Molnar, C. et al. in xxAI – Beyond Explainable AI (eds Holzinger, A. et al.) 39–68 (Springer, 2022).