GSEA原理和使用方法浅析
在RNA-seq数据的分析流程中,基因集富集分析(Gene Set Enrichment Analysis,GSEA)是一种很常用的下游分析方法。这一方法在2005年由MIT博德研究所开发,用于确定先验定义的基因集(如特定信号通路的编码基因、疾病性状的关联基因等)是否在基因表达差异分析的结果上出现统计学显著性。
因为课题的原因,最近再一次复习了一下GSEA方法。目前有许多工具都能进行GSEA分析,例如博德研究所官方提供了一个基于Java编程语言开发的GSEA计算工具 ,南方医科大学的余光创教授开发的R语言工具包ClusterProfiler也提供了GSEA分析的功能。本文将简单介绍一下GSEA的原理,并举例说明如何使用博德研究所官方的工具包做GSEA分析。
一、原理
正如前文所述,GSEA想看的是一系列基因集在差异分析结果中的显著性。以下图为例,其来自于Cell期刊上一篇研究疾病模型的文章( Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis)。在这幅图中,作者想要研究GATA基因的突变对心脏功能的影响,作者首先在GATA基因野生型(iwt)和突变型(G296S)两组样本之间进行了差异表达分析,找到了一系列差异基因,随后用不同的基因集做GSEA,发现Cardiac Development(心脏发育)基因集在野生型中普遍表达偏高,而Endothelial/endocardial(内皮或内膜发育)基因集在突变型中表达偏高,因此推测GATA基因的G296S突变可能通过心脏内皮或内膜的过度发育导致心脏相关疾病的产生。
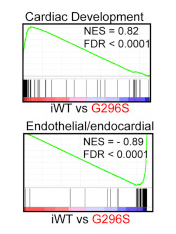
我们可以观察一下这幅图。在每个小图中,都可以分为上下两个部分,上半部分是一条绿色的曲线,其与y=0轴围成的曲线的面积代表GSEA的富集分数,这个分数的正负号代表基因集富集的方向,而绝对值反映了基因集在我们的差异基因列表中是否显著富集。下半部分是一系列黑线,黑线的位置代表了基因集在我们的差异基因列表中的位置。GSEA的原理就是看一个特定基因集(如果不懂什么叫基因集,可以简单理解为一个GO term或KEGG pathway)在我们实验得到的gene rank list中富集到的位置。一般来说,gene rank list会按照差异表达进行排序,越靠前说明表达差异越大;而一个基因集如果富集到gene rank list中更加靠前的位置,则说明这个基因集在我们的实验中更重要。
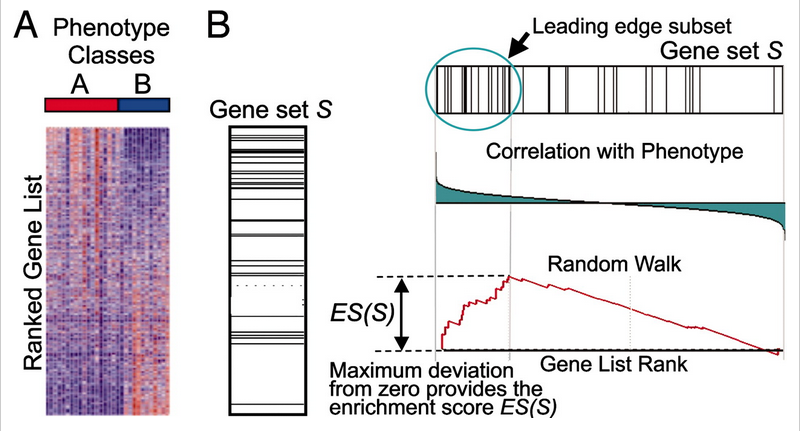
更直观的原理可以参考上面这张图。首先,给定一个按基因表达差异排序的基因表 $L$ 和一个预先定义的基因集 $S$ ,GSEA的目的是判断 $S$ 里面的成员 $s$ 在 $L$ 里面是随机分布还是聚集在 $L$ 的某一侧(顶部或底部)。若研究的基因集 $S$ 的成员显著聚集在 $L$ 的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。主要步骤包括下面三步:
- step1:富集分数(ES)的计算。ES反映了一个 $S$ 在整个 $L$ 的某一侧(顶部或底部)过度表达的程度。计算得分的方法是沿着 $L$ 向下走,每走一步时,如果遇到 $S$ 中的一个基因,则
running-sum统计量增加,否则running-sum统计量减少,增加或减少的幅度取决于基因的权重。最终的running-sum会形成一条曲线,如本文第一张图中的绿色曲线和上图中右下角的红色曲线。 - step2:估计ES的显著性。这里使用了置换检验的方法进行统计检验。
- step3:对显著性p值的矫正。通常我们在做GSEA时一次会测试若干个基因集 $S$ 在 $L$ 中的富集情况,因此需要使用FDR等方法矫正p值。
一次GSEA会检测几百甚至几万个基因集。这些基因集可以由研究者自行定义,也可也直接使用MSigDB数据库预先定义的基因集。在MSigDB数据库中,这些基因集被归类为H、C1、C2等多个大类,研究者可以一次选择所有的基因集做GSEA,也可只挑选其中一部分感兴趣的基因集。一般来说,选择C1基因集则是看基因的染色体区域分布,C5基因集则是GO term的富集分析。

关于GSEA和MSigDB的更多内容,还可以参考官方文档
二、安装
GSEA的安装包可以从官方网站的下载界面 https://www.gsea-msigdb.org/gsea/downloads.jsp 中下载。首次打开这个页面需要注册(如下图),注册步骤很简单,不需要等待太久。
注册完成后登录,就可以看到安装包的下载链接了。如下图,GSEA提供了多个不同平台的安装包版本,其中前三个自带了java11的运行环境,一键安装即可,而最后一个跨平台的命令行版本需要使用系统自带的java。如果怕麻烦,直接下载前三个安装包就好了。
除了GSEA软件本体,我们还需要下载预定义的基因集。MSigDB提供了基因集的下载链接,见 https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp 。如下图,这里提供了好几种格式的基因集压缩包,其中包括gene symbol版的gmt文件、NCBI Enterz版的gmt文件,以及sqlite数据库格式和json格式。做GSEA的话我们只需要前面两种,具体使用gene symbol版还是NCBI Enterz版要看具体情况。
三、数据格式
GSEA需要的文件格式包括下面这几类。
| Data File | Content | Format | Source |
|---|---|---|---|
| Expression dataset | Contains features (genes or probes), samples, and an expression value for each feature in each sample. Expression data can come from any source (Affymetrix, Stanford cDNA, and so on). | res, gct, pcl, or txt | 差异表达矩阵文件 |
| Phenotype labels | Contains phenotype labels and associates each sample with a phenotype. | cls | 自行提供,需要与表达矩阵中的样本一一对应起来 |
| Gene sets | Contains one or more gene sets. For each gene set, gives the gene set name and list of features (genes or probes) in that gene set. | gmx, gmt or grp | 预定义的基因集文件。MSigDB中提供了这一类文件的下载,也可也根据官方文档自行定义 |
| Chip annotations | Lists each identifier on a platform and its matching HGNC gene symbol. Optional for the gene set enrichment analysis. | chip | 芯片探针数据的转换文件。可选。 |
| Ranked List File Format | It is used when you have a pre-ordered ranked list that you want to analyze with GSEA. | rnk | 如果使用GSEAPreranked模式,则需要提供这个文件 |
各种文件格式的定义方式都可在官方文档中查看
四、在Linux命令行中使用GSEAPreranked模式进行GSEA分析
因为我的实验数据并非基因差异分析数据,无法提供差异表达矩阵,只能使用GSEAPreranked模式跑分析。首先我们要准备一下.rnk文件(文件名为data.rnk),部分内容如下:
1 | EXD1 -0.5544303794287568 |
.rnk文件是一种制表符分隔的文本文件,第一列是基因名称,第二列是基因的权重。在这个文件里面,基因排序与否并无所谓,因为GSEA会根据基因权重再做一次排序。
我们还需要一份预定义的基因集文件msigdb.v2023.1.Hs.symbols.gmt。这个文件从MSigDB网站直接下载即可。
使用下面这份bash脚本,即可完成对我们的结果的GSEA检验。其中调用的是gsea-cli.sh这个脚本,部分传入参数的含义在下面进行了解释。
1 |
|
每一次运行会建立一个文件夹,并在文件夹内生成大量结果文件(html+csv+png),其中index.html是概括性的文件,部分信息如下。

可以看出这里列出了使用到的geneset的数量和富集显著的geneset的数量;除此之外,点击链接可以看到更详细的显著富集的geneset的名称和p-value,以及图片报告,如下:
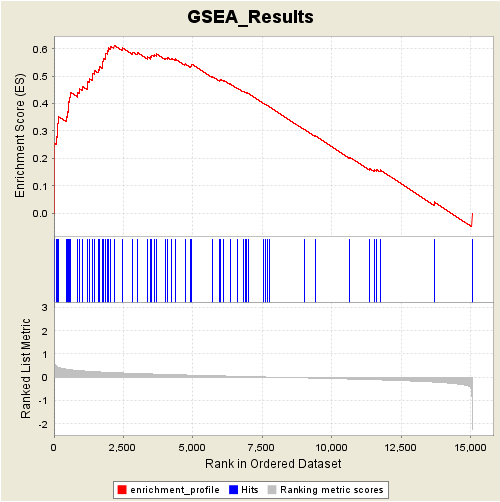
蓝色竖线代表geneset在gene rank list中出现的位置,如果蓝色竖线越在左侧集中,则说明这个geneset富集程度越高。
五、其他补充 & 参考文献
当然,博得研究所开发的这款GSEA软件,也支持图形界面下的操作,甚至可以和cytoscape联动绘制不同基因集之间的相互作用。这一部分的内容我没有做太多深究,想要进一步了解的朋友可以参考 《一文掌握GSEA通路富集分析,超详细教程!》 - 生信宝典 陈同的文章 - 知乎 。本文其他参考文献如下:
- 《手把手教你用R做GSEA分析》 - 生信小课堂的文章 - 知乎
- 《差异基因没办法富集到通路你就放弃了吗》 - 曾健明的文章 - 知乎
- 《GSEA法基因功能富集分析原理详解》- 组学大讲堂的文章 - PLoB
其他链接: