rGREAT:适用于R语言的GREAT工具
上一篇博客中,我们介绍了GREAT这个基因组注释工具,以及它的网页版界面的使用方法。在本篇博客中,我们将更深入的介绍rGREAT——这个工具的R语言版本,并展示一些高级用法。
一、什么是rGREAT
rGREAT 是一个 R/Bioconductor 包,实现了 GREAT(Genomic Regions Enrichment of Annotations Tool) 算法,用于对基因组区域进行功能富集分析。与传统的基因集富集分析不同,GREAT 直接将基因组区域(如ChIP-seq峰、ATAC-seq开放区域等)与基因功能注释关联起来,从而揭示这些区域可能参与的生物学过程、通路或功能类别。
使用场景:
- ChIP-seq 转录因子结合位点的功能注释
- ATAC-seq 或 DNase-seq 开放染色质区域的功能分析
- 差异甲基化区域的功能富集分析
- 任何基因组区间集的功能解读
二、安装方法
(一)通过命令行安装rGREAT本体
有三种方法可以安装rGREAT,任选其一即可:
- rGREAT 可通过 Bioconductor 安装(在R-session中运行):
1 | if (!requireNamespace("BiocManager", quietly = TRUE)) |
- 也可从 GitHub 安装最新开发版(在R-session中运行):
1 | library(devtools) |
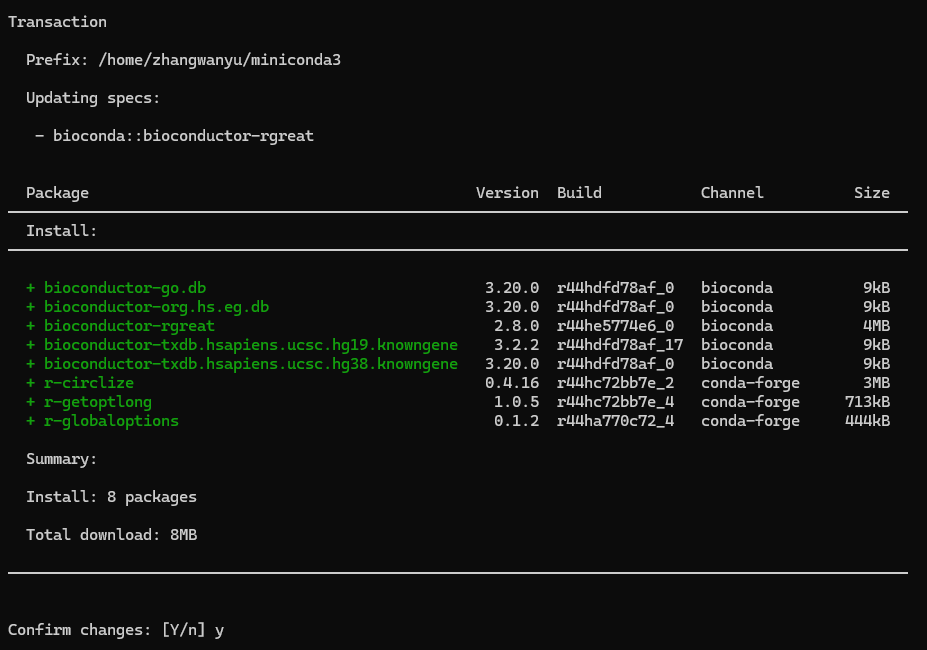
- 还可以通过conda或者mamba安装(在bash或cmd等终端中运行):
1 | conda install bioconda::bioconductor-rgreat |
(二)Trouble-shooting:如果依赖项安装出错,怎么办
有一些附带的包(例如 GO.db )会一并安装。如果在上面的流程中这些依赖项安装出错,可以使用下面的指令在R-session中进行安装:
1 | ## 首先切换软件源。 |
如果能够使用 library(rGREAT) 指令成功加载这个软件包(产生如下图所示的输出),则说明安装成功。
三、rGREAT 的使用方法
主要函数概览:
submitGreatJob():提交在线分析great():执行本地分析getEnrichmentTable():获取富集结果getRegionGeneAssociations():获取区域-基因关联信息plotRegionGeneAssociations():可视化区域与基因的关系shinyReport():启动交互式 Shiny 报告
rGREAT 支持两种分析方式:
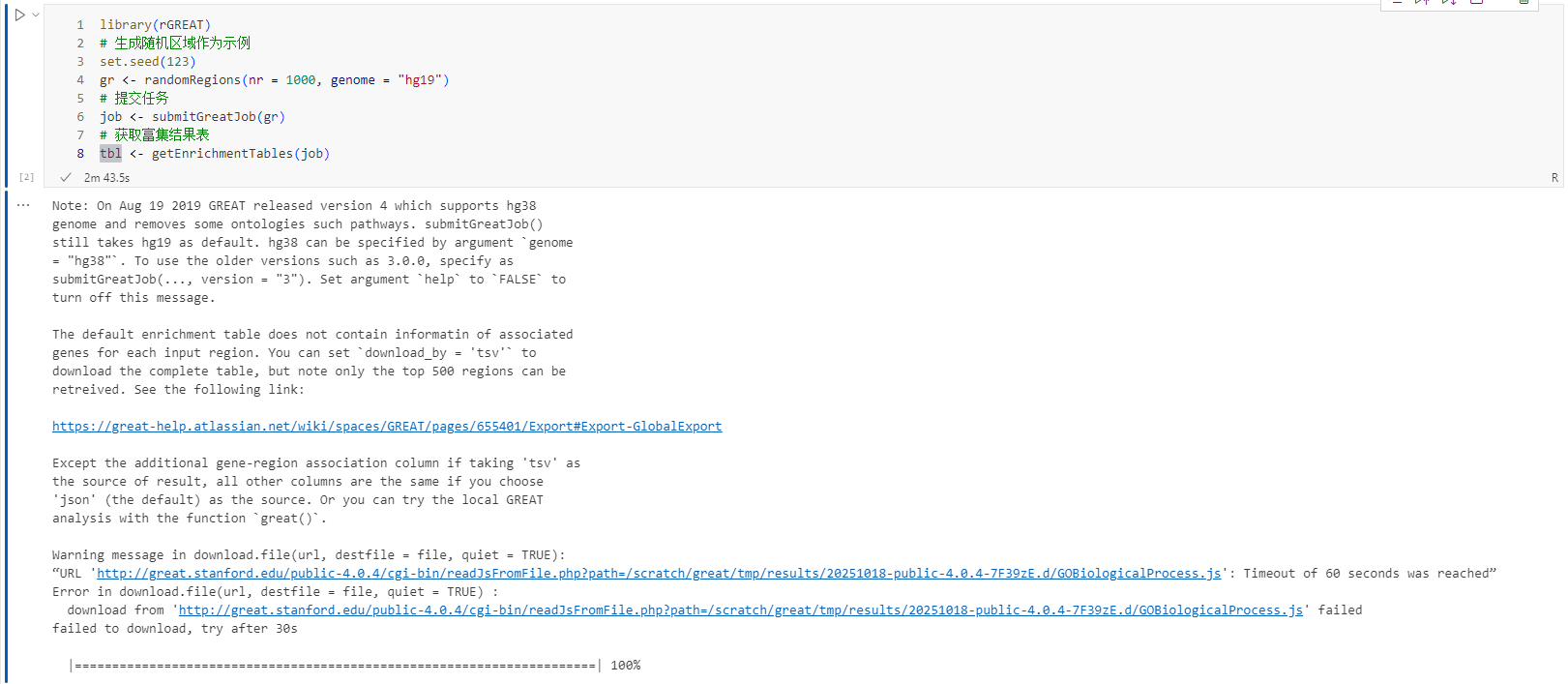
1. 在线分析(Online GREAT Analysis)
将区域提交至 GREAT 官方网站,获取标准化结果。
1 | library(rGREAT) |
输出:
2. 本地分析(Local GREAT Analysis)
使用内置的 GREAT 算法在本地执行分析,支持自定义基因集和物种。
本地分析的优势:
- 支持600+物种
- 可使用自定义基因集
- 无需网络连接
- 灵活设置背景区域
1 | res <- great(gr, "MSigDB:H", "TxDb.Hsapiens.UCSC.hg19.knownGene") |
输出:
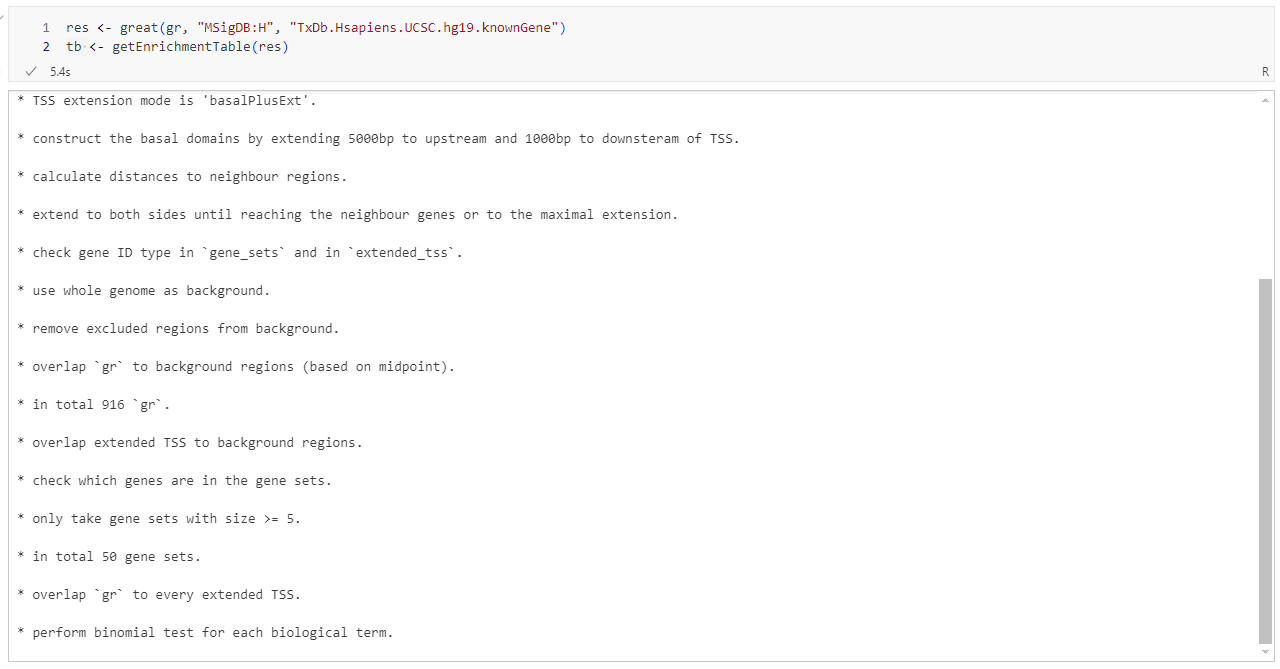
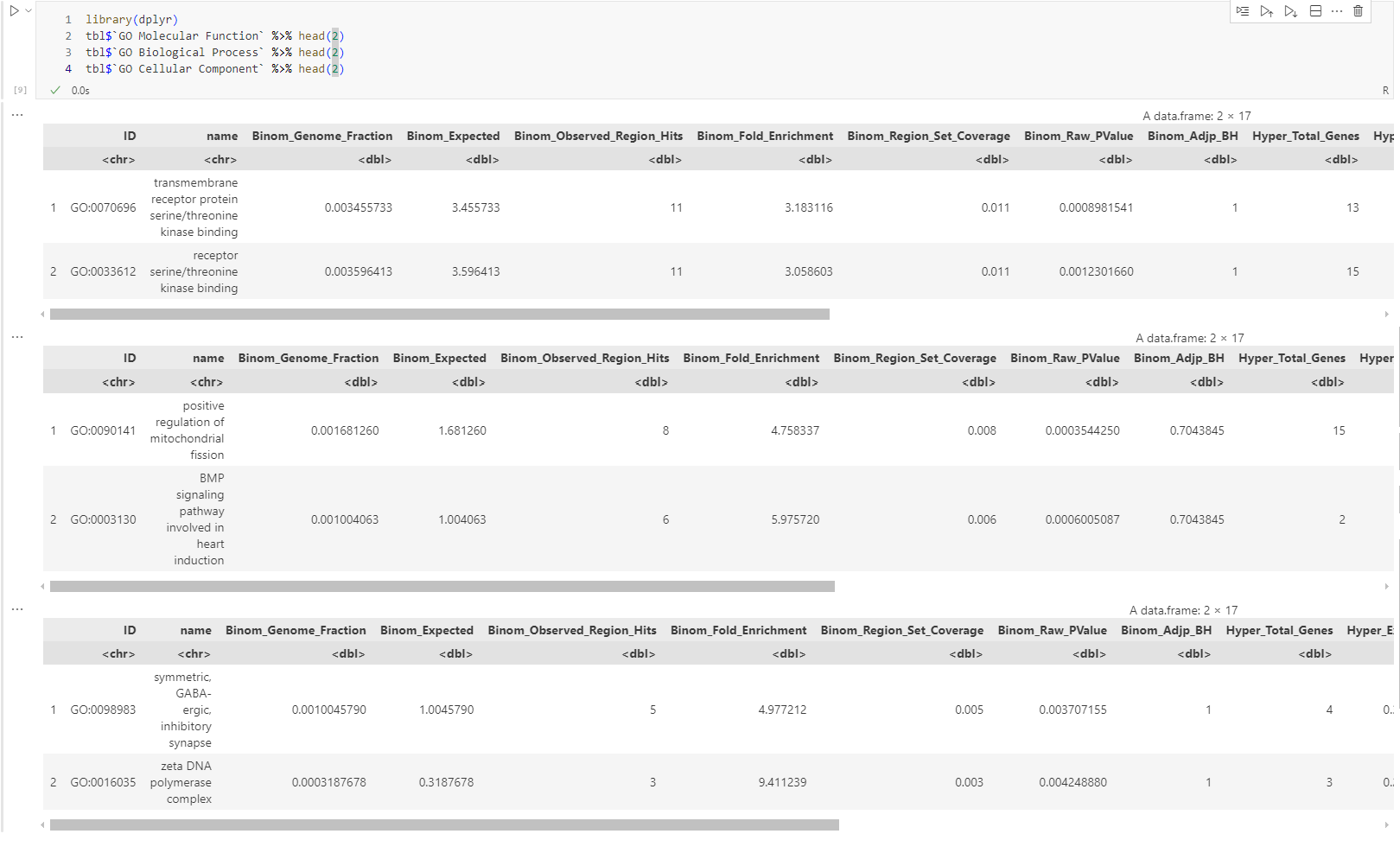
五、结果解读与可视化
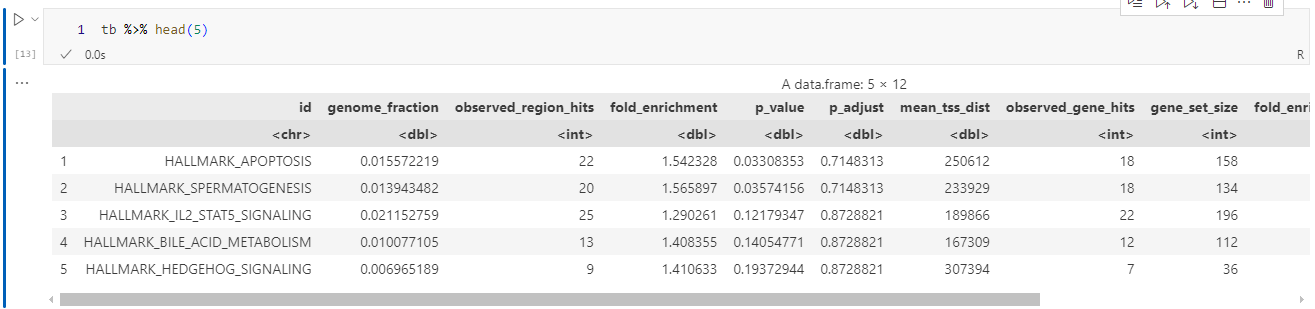
富集结果通常包含以下列:
ID:功能术语ID(如GO:0004984)name:术语名称p_value/p_adjust:原始/校正后的 p 值fold_enrichment:富集倍数region_hits:输入区域中与该术语相关的区域数gene_hits:与该术语相关的基因数
rGREAT 提供多种可视化方法:
- 区域-基因关联图
- 距离TSS的分布图
- 火山图(Volcano plot)
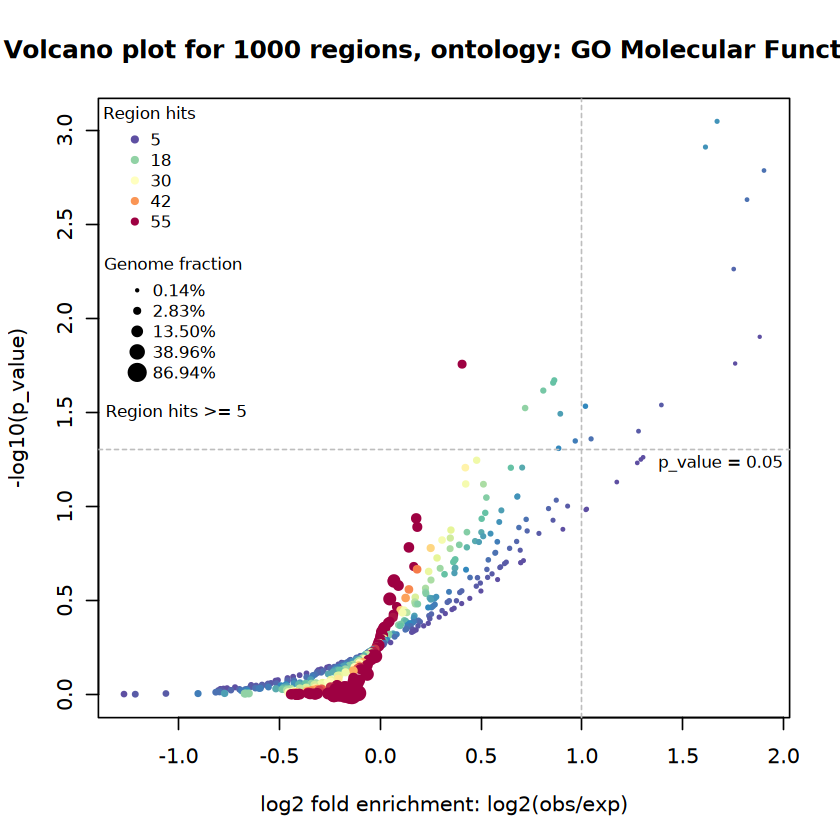
火山图
我们可以对GreatJob或GreatObject对象使用plotVolcano()来绘制火山图,直观地展示富集最显著的条目 。
1 | #library(rGREAT) |
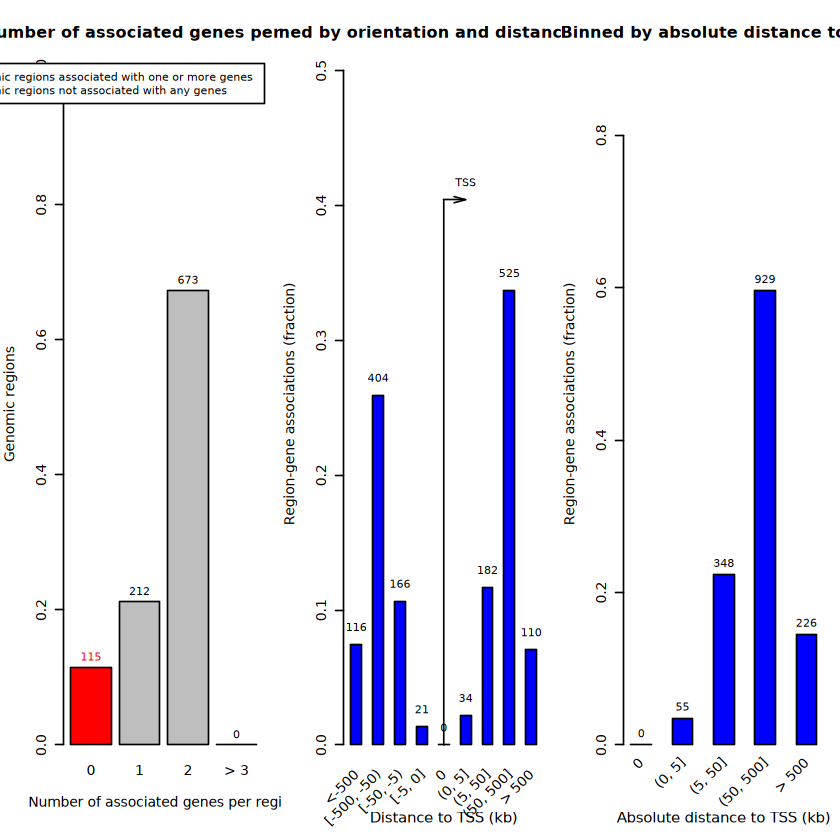
区域-基因关联图 (plotRegionGeneAssociations)
这是GREAT的特色图。它可以展示输入区域与基因TSS的距离分布 ,以及区域与基因的关联情况 。
1 | #library(rGREAT) |
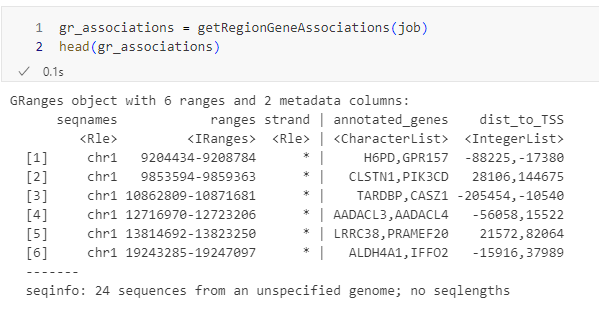
获取关联数据 (getRegionGeneAssociations)
如果我们想获得区域到基因的具体关联信息(例如哪个区域关联到了哪个基因,距离多远),可以使用getRegionGeneAssociations() 。
1 | #library(rGREAT) |
交互式shiny文档报告
rGREAT还提供了一个shinyReport()函数,它可以在本地启动一个Shiny应用,让我们以交互方式探索和筛选富集结果 。
1 | #library(rGREAT) |
六、总结与参考文献
rGREAT是一个强大且灵活的R包,它将GREAT算法的分析能力带入了R/Bioconductor生态。无论是想快速调用在线服务,还是需要利用本地模式对非模式生物进行复杂的自定义注释分析,rGREAT都能提供完善的解决方案。
如果读者朋友正在处理基因组区域数据,并希望探索它们的功能意义,rGREAT无疑是一个值得尝试的工具。
以上。
参考文献以及一些有用的链接:
参考:
- Zuguang Gu, et al., rGREAT: an R/Bioconductor package for functional enrichment on genomic regions. Bioinformatics, 2023.