GREAT:一种富集分析与可视化的方法
基因组区域富集注释工具(Genomic Regions Enrichment of Annotations Tool, GREAT)是一个用来进行功能注释和下游分析的工具集,用于分析通过全基因组范围内 DNA 结合事件的局部测量所确定的顺式调控区域的功能意义。与传统的基因富集分析工具如DAVID、enrichr或clusterprofiler不同,GREAT直接分析基因组区域而非基因列表,能够更全面地评估非编码区域的功能意义。
和先前的方法(如DAVID、enrichr等基于基因集列表的方法)相比,GREAT方法考虑了基因组片段,也能够正确纳入远端结合位点,因此在研究基因调控关系等方面效果更好。
一、计算原理(by AI总结)
参考论文:McLean, C., Bristor, D., Hiller, M. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501 (2010). DOI: 10.1038/nbt.1630
GREAT 的基因组注释原理
GREAT (Genomic Regions Enrichment of Annotations Tool) 的核心创新在于它直接对“基因组区域”(genomic regions) 而非“基因列表”(gene list) 进行功能富集分析。它解决了传统方法在处理远端顺式调控元件(如增强子)时的两大根本性缺陷:信息丢失和假阳性偏差。
其原理可以分解为以下三个关键步骤:
定义“调控域”(Regulatory Domain) 而非简单关联:
- 传统的“最近基因法”或“2kb内”方法非常武断,会丢失大量远端调控事件。
- GREAT 提出一个更符合生物学实际的模型:为基因组中的每一个基因定义一个“调控域”。这个域不仅包括启动子区域(默认5kb上游 + 1kb下游),还允许向两侧延伸,直到遇到邻近基因的调控域边界,但最大不超过1Mb(用户可自定义参数)。这模拟了基因可能受到其上下游一定范围内所有调控元件影响的概念。
- 每个输入的基因组区域(例如ChIP-seq峰)会被映射到其覆盖的所有基因的调控域中。这意味着一个区域可以同时关联多个基因。
采用“二项检验”(Binomial Test) 作为统计基础:
- 这是GREAT最核心的统计学创新。它不计算有多少“基因”被选中,而是计算有多少“基因组区域”落在了具有特定功能注释的基因的调控域内。
- 具体来说,它首先计算整个基因组中,被特定功能术语(如“细胞骨架”)所覆盖的基因的调控域总长度占全基因组的比例(
p_π)。这个比例就是该功能术语的“先验概率”。 - 然后,它使用二项分布来检验:观察到有
k个输入区域落在这些“功能相关”的调控域内,在总共n个输入区域的情况下,是否显著多于随机期望值(即n * p_π)。 - 关键优势:这种方法天然地校正了“调控域大小偏差”。那些调控域很大的基因(如发育相关基因),其调控域在基因组中占比就大,因此随机情况下也更容易捕获到调控区域。二项检验通过
p_π将这种大小差异纳入考量,从而避免了将这些大基因相关的通用功能错误地判断为显著富集(假阳性)。
结合多种本体论并提供双重验证:
- GREAT整合了20种不同的本体论数据库,涵盖基因功能(GO)、表型、疾病、通路、表达谱、转录因子靶点、基因家族等多个维度,提供了丰富的注释资源。
- 它同时运行二项检验(基于区域)和传统的超几何检验(基于基因),并将结果对比展示:
- 仅二项检验显著:可能代表少数几个关键基因吸引了大量调控区域(基因特异性富集)。
- 仅超几何检验显著:很可能是由调控域大小偏差导致的假阳性。
- 两者均显著:这是最可靠的结果,表明该功能不仅在区域层面富集,而且涉及的基因数量也足够多,提供了强有力的支持。
GREAT 与其他工具的不同之处
如下图,是文章中对现有方法(a图)和GREAT方法(b图)的方法总结与对比。GREAT方法使用二项分布进行建模,计算基因组区域的覆盖长度(而传统方法使用超几何分布,计算基因集的交集),因此可以更好的分析调控元件的相互作用。

| 特征 | GREAT | DAVID / clusterProfiler / Enrichr |
|---|---|---|
| 分析对象 | 基因组区域 (Genomic Regions)。输入是一系列坐标区间(BED文件)。 | 基因列表 (Gene List)。输入是一个基因ID列表(如从DEG分析得到)。 |
| 关联方式 | 基于调控域模型(如5+1kb + 最大1Mb扩展)。一个区域可关联多个基因。 | 主要基于距离。通常是取峰附近最近的基因,或限定在TSS ± X kb内的基因。一个区域通常只关联一个或两个最近的基因。 |
| 核心统计模型 | 二项检验 (Binomial Test),以基因组覆盖率为背景。显式地校正了调控域大小带来的偏差。 | 超几何检验 (Hypergeometric Test) 或 Fisher精确检验。以基因总数为背景。无法校正调控域大小偏差,易产生假阳性。 |
| 主要优势 | 1. 充分利用远端调控信息,避免因忽略远端峰而丢失重要功能。 2. 有效控制假阳性,特别是对于调控域大的基因。 3. 分析结果更贴近真实的顺式调控机制。 |
1. 应用范围广,适用于任何能生成基因列表的场景(如RNA-seq, microarray)。 2. 生态成熟,拥有庞大的数据库支持和丰富的可视化功能(尤其是clusterProfiler)。 3. 操作简便,流程标准化。 |
| 主要局限/适用场景 | 专用于顺式调控数据。必须有明确的基因组坐标(如ChIP-seq, ATAC-seq, ChIA-PET等)。不能直接用于分析基因表达水平变化。 | 不适合分析富含远端调控元件的数据。当输入的调控区域大部分位于基因远端时,这些方法会严重低估或完全遗漏其功能。 |
总结来说:
- DAVID、clusterProfiler、Enrichr 是经典的基因列表富集分析工具,它们的输入是“哪些基因发生了变化”,输出是“这些基因参与了什么功能”。它们依赖于“调控=靠近基因”的简化假设。
- GREAT 是专门为顺式调控区域富集分析设计的下一代工具。它的输入是“基因组上哪些区域有功能活性”,输出是“这些活性区域的功能意义是什么”。它通过建立“调控域”模型和使用“二项检验”,打破了“调控=靠近”的限制,能够更准确、更全面地解读远端调控元件的生物学功能,尤其是在研究增强子、超级增强子等长距离调控机制时,GREAT的优势尤为突出。
二、使用方法(网页端接口)
此处需要访问GREAT的官网: http://great.stanford.edu/public/html/
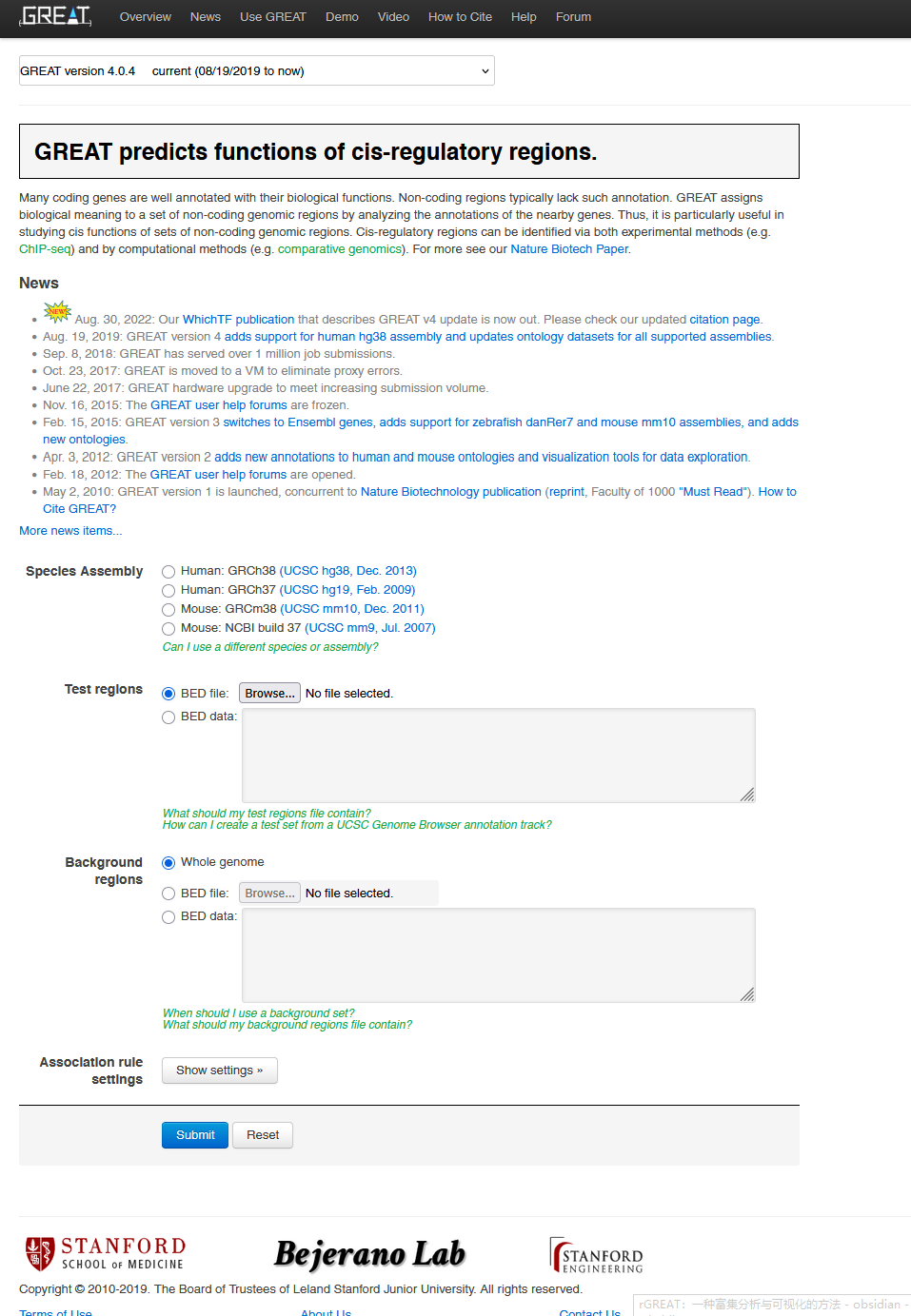
与DAVID等网站的使用方法不同,GREAT在注释时需要准备一个用于检验的bed文件,即test region文件。如有需要,可以再准备第二个背景区域的文件(background region),其必须是test region所包含区域的超集。
关于背景区域文件,官方给出的说明如下图(更多详情见网站)。这个文件是可选的,如果不上传,则在统计检验中会使用全基因组的所有区域作为检验区域。

所谓bed文件,就是以tsv表格形式存储的基因组坐标,其中第一列是染色体编号,第二列是基因组区域的起始位置,第三列是基因组区域的终止位置,从第四列开始用户可以存储一些自定义的信息(例如一些元数据)。下面是一个bed文件的例子:
1 | chrX 55009055 55030977 ALAS2 |
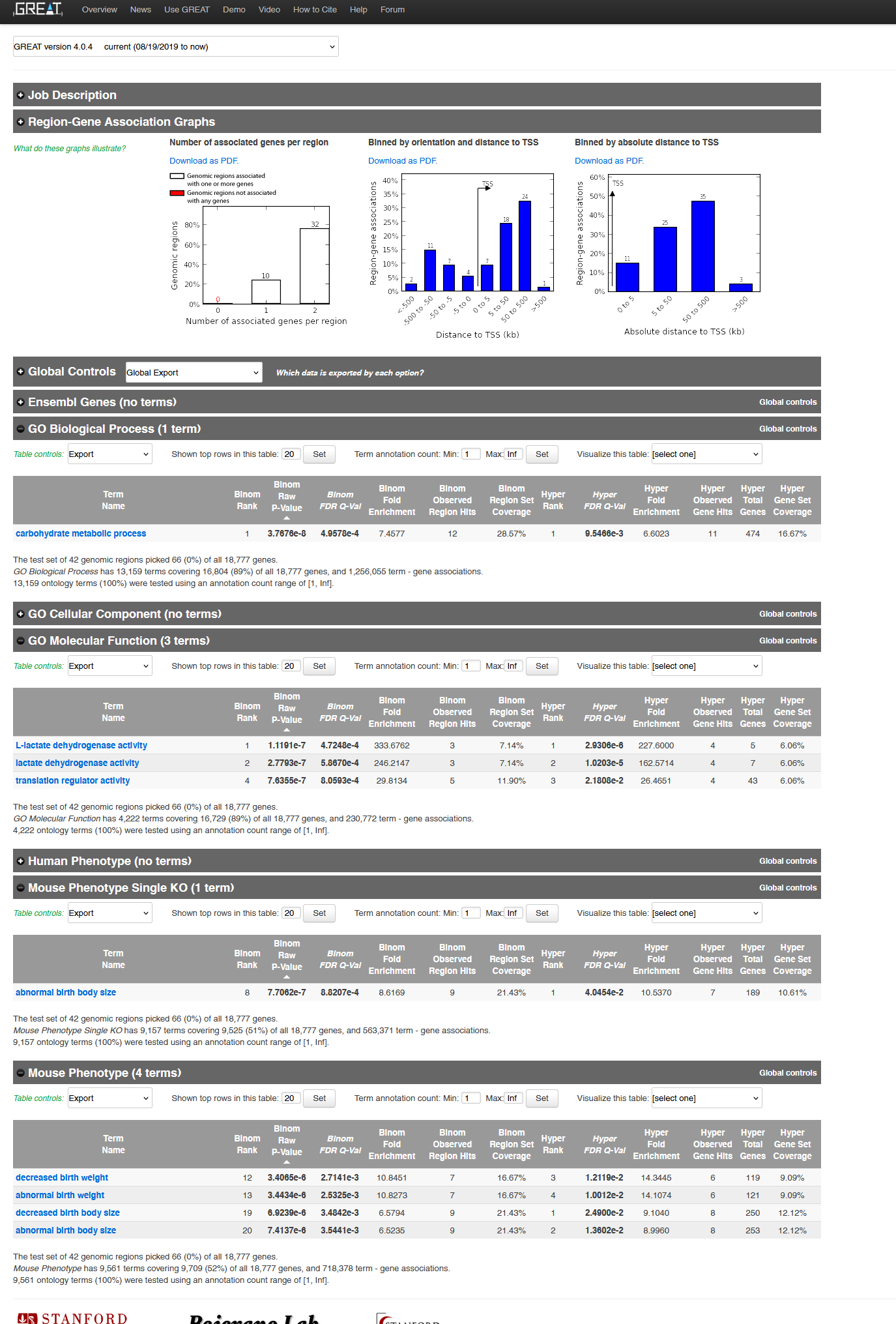
上传bed文件,选择合适的物种参考基因组(例如上面那个bed文件使用的是人类GRCh38的参考基因组坐标),然后点击submit就可以运行了。输出的结果如下所示:
三、附带小工具:一个从gtf文件转bed文件的python代码
这个工具是在组会上听到的,笔者在自己的课题中暂时还没有涉及到需要对基因组调控区和非编码区做功能注释的工作。为了能够检验这个工具的效果,笔者攒了一个小工具,通过给定基因列表和GTF文件,来得到一份bed格式的test region文件( result.bed )。以下是代码:
1 | import pandas as pd |
四、下期预告:基于R包rGREAT的注释
rGREAT是一个基于GREAT方法的R包,其功能更全面,可以自定义的选项也更多。
本来应该这期一起讲的,但是来不及准备了。所以,相关内容留到下期,敬请期待!
一些有用的参考链接: