基于crux的蛋白质谱定量
在近期工作当作,我需要处理一个蛋白质组学的数据集,于是在数据处理和查询资料的过程中,学习了相关的知识,以及crux的用法。
1、背景
1.1 蛋白质组学的测序原理和数据来源
蛋白质组学研究的测序主要采取质谱方法。质谱方法的测序原理略(下面是一个AI总结)。
质谱法的基本原理是将样品分子电离,并按照其质荷比(m/z)进行分离和检测。蛋白质组学中,蛋白质首先被酶切(如胰蛋白酶)分解成小肽 段,然后通过液相色谱(LC)将这些肽段分开,再通过质谱仪进行分析 。
质谱仪主要有两种类型:串联质谱(MS/MS)和飞行时间质谱(TOF)。 在串联质谱中,首先使用一级质谱(MS1)对肽段进行初步的质量分析,然后选择特定质量的肽段进行碰撞诱导解离(CID),产生二级碎片离子,最后通过二级质谱(MS2)对这些碎片离子进行质量分析。通过对这些碎片离子的质量进行比较,可以推断出原始肽段的序列信息。而在飞行 时间质谱中,样品分子在电场作用下加速,根据它们的质荷比不同,以 不同的速度穿过真空管道,从而实现分离和检测。
通过比较实验获得的质谱数据与理论数据库中的肽段信息,可以确定蛋 白质的身份和修饰状态,进而揭示整个蛋白质组的组成和变化。
事情是这样的,在近期工作当作,我需要处理一个蛋白质组学的数据集。这个数据集包含了若干raw文件(质谱平台的原始下机数据)、若干mgf文件(质谱峰列表文件)和一个mzid文件(质谱鉴定结果)。我需要将它们整理为表达矩阵的格式。但是mgf文件和mzid文件都不是我熟悉的文件类型,于是进行了一番探索(特别是后者)。
1.2 MZID格式介绍
参考:
质谱数据文件解析(一)—mzML格式 - 馒头的文章 - 知乎
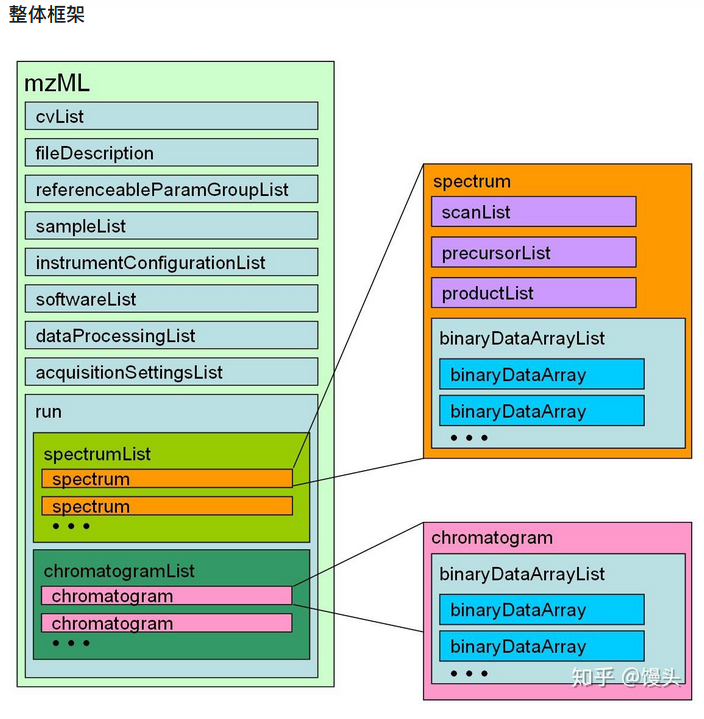
这是Institute for Systems Biology (ISB) 提出的一种质谱数据存储格式,旨在统一之前充斥于市面上的mzData和mzXML两种不同的数据格式。其本质上是一个XML文件,通过各种标签存储谱图信息。
2、MZID的处理工具
2.1 PEAKS Studio
参考:
但是这个工具是商业软件,且测试版国内下载很不方便(网站上说 “It appears that you are from Mainland China. Baizhen Biotechnologies is our distributor in this region. Please send email to sales-china@bioinfor.com or phone +86-21-60919891 to request your demo.”。给提到的国内代理商发邮件,并没有得到回信)。遂放弃探索。
2.2 crux
后来,检索到了一篇论文:
Jones, Andrew R et al. “The mzIdentML data standard for mass spectrometry-based proteomics results.” Molecular & cellular proteomics : MCP vol. 11,7 (2012): M111.014381.
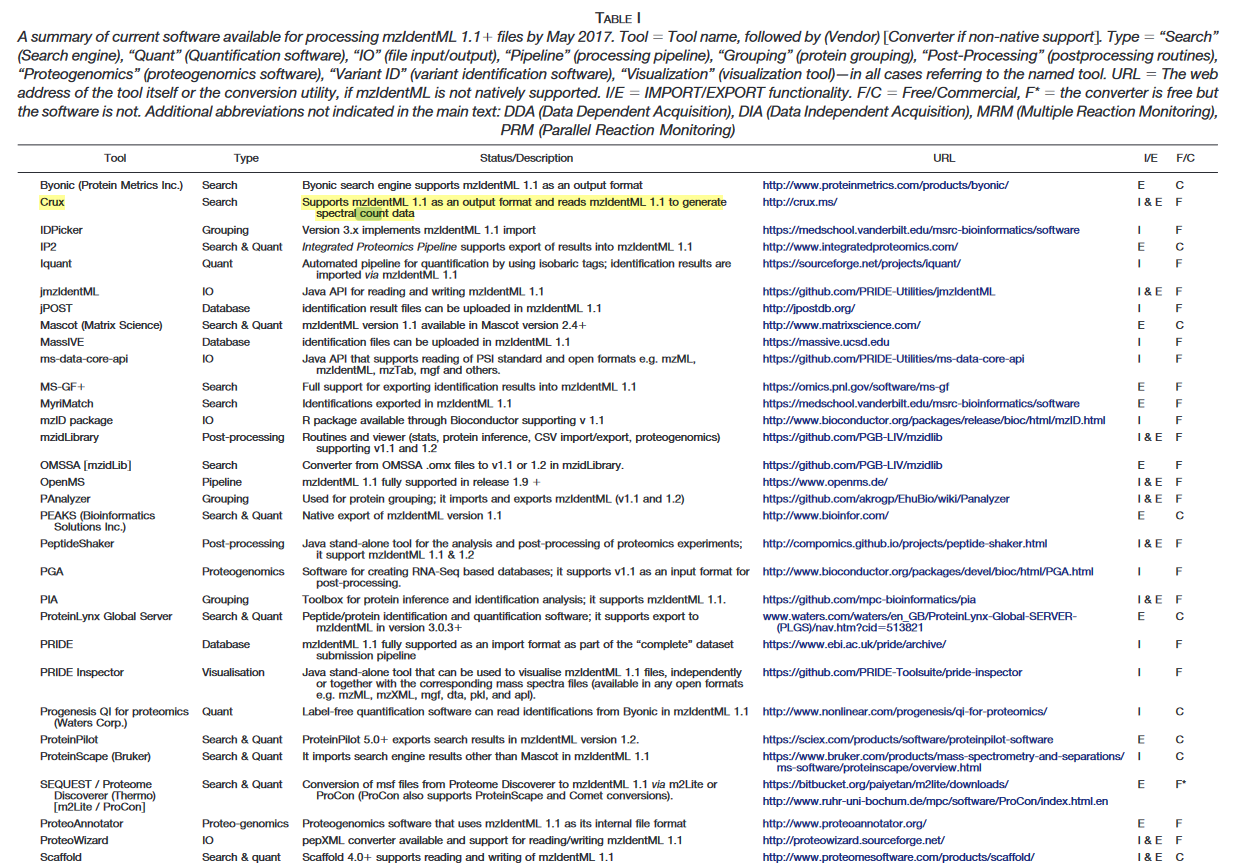
这篇论文中提到了crux这个开源工具,可以读取mzid格式的蛋白质谱,并进行定量分析。这是我们需要的。
3、基于crux的蛋白质谱定量
3.1 安装
在crux官网下载页面上下载对应的系统版本的包即可。
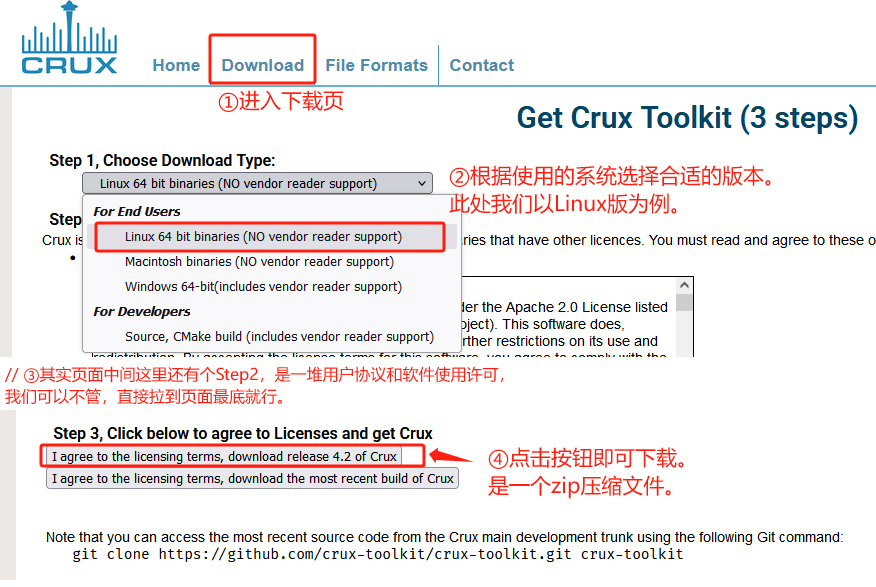
下面我们以Linux为例说一说使用方法。
假设我们下载的文件名称是 crux-4.2.Linux.x86_64.zip ,现在我们用下面的指令进行解压,然后进入软件目录,就可以看到文件结构
1 | unzip crux-4.2.Linux.x86_64.zip -d crux |
整个目录的结构如下:
1 | . |
其中,在 bin 目录下有一个单一的可执行文件 crux ,这个就是主程序。
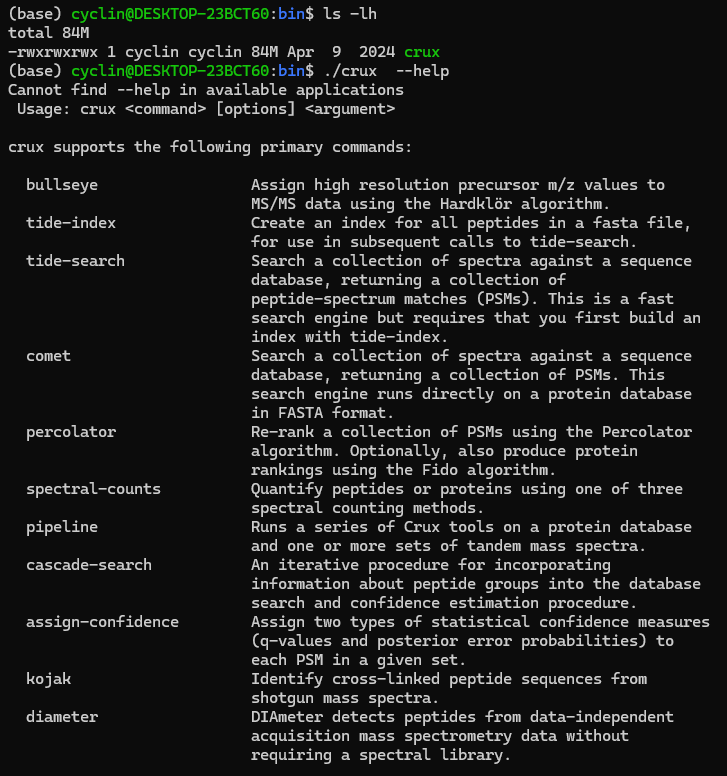
我们也可以把crux所在的目录加入系统环境变量,以便于后面我们用 crux 指令进行访问。
3.2 crux的使用方法(转码和定量)
参考crux官方文档:
PSM文件代表肽段-谱图匹配(Peptide-Spectrum Match)文件,通常包含每个质谱谱图对应的肽段信息、得分、q值等。PSM文件可以是不同的格式,包括mzid格式、SQLite格式、tsv格式、pepXML等
我们的谱文件是mzid格式的。问了一下DeepSeek,其建议我先转换为tsv格式,以进行下一步的处理。
1 | # 我们的输入文件名是peptides_1_1_0.mzid.gz |
上述步骤将会在当前目录创建文件夹 crux-output ,然后在这个文件夹中生成三个文件:
1 | psm-convert.log.txt |
其中的 psm-convert.txt 是转码结果。
随后的定量过程需要 crux spectral-counts 指令。定量过程还需要准备fasta格式的数据库文件,见下一步。
3.3 在uniprot上下载对应的fasta文件的方法
假设我们测序的物种是人(HUMAN)。我们按照下图中的步骤,完成这一文件的下载。
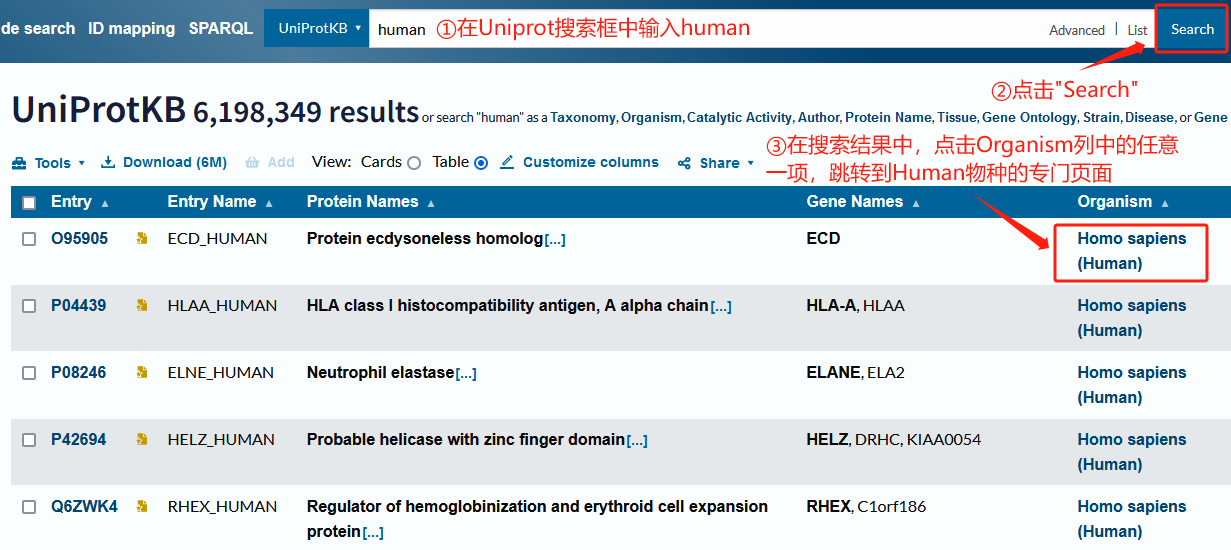
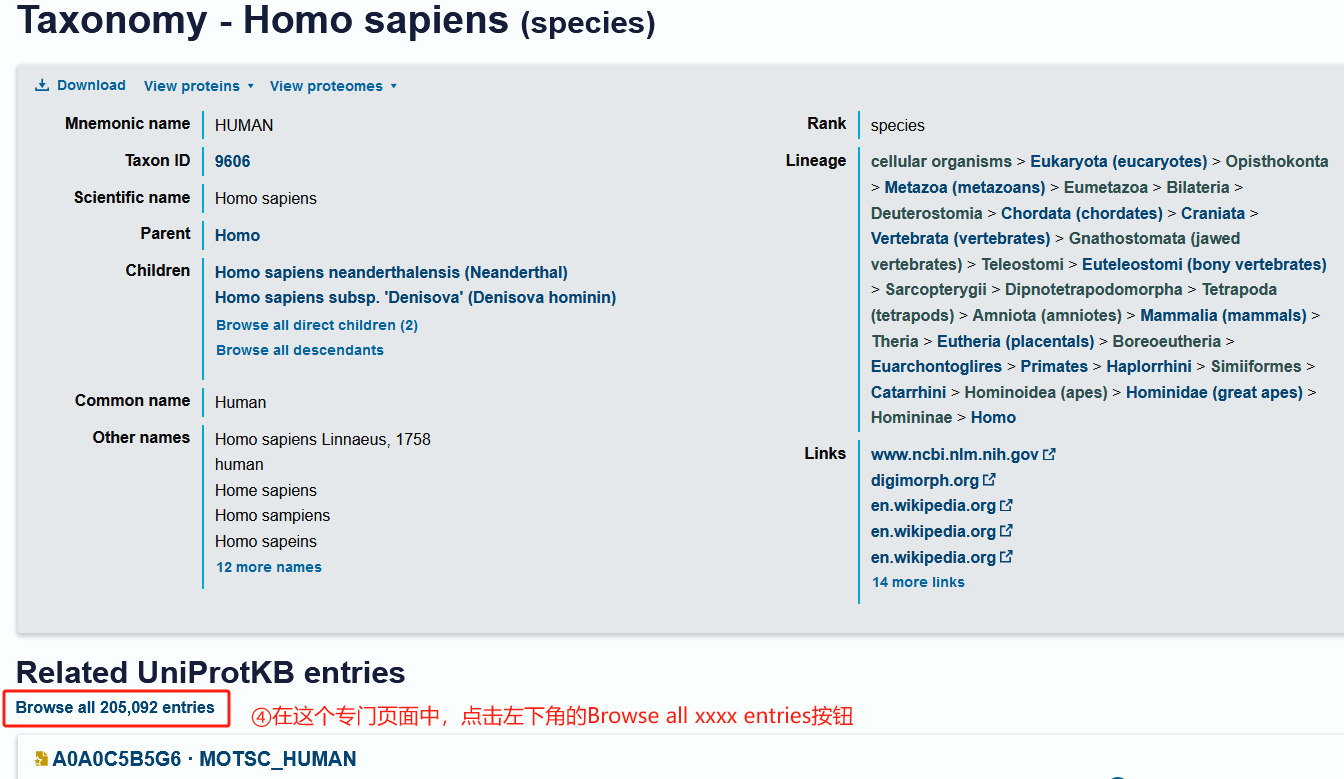
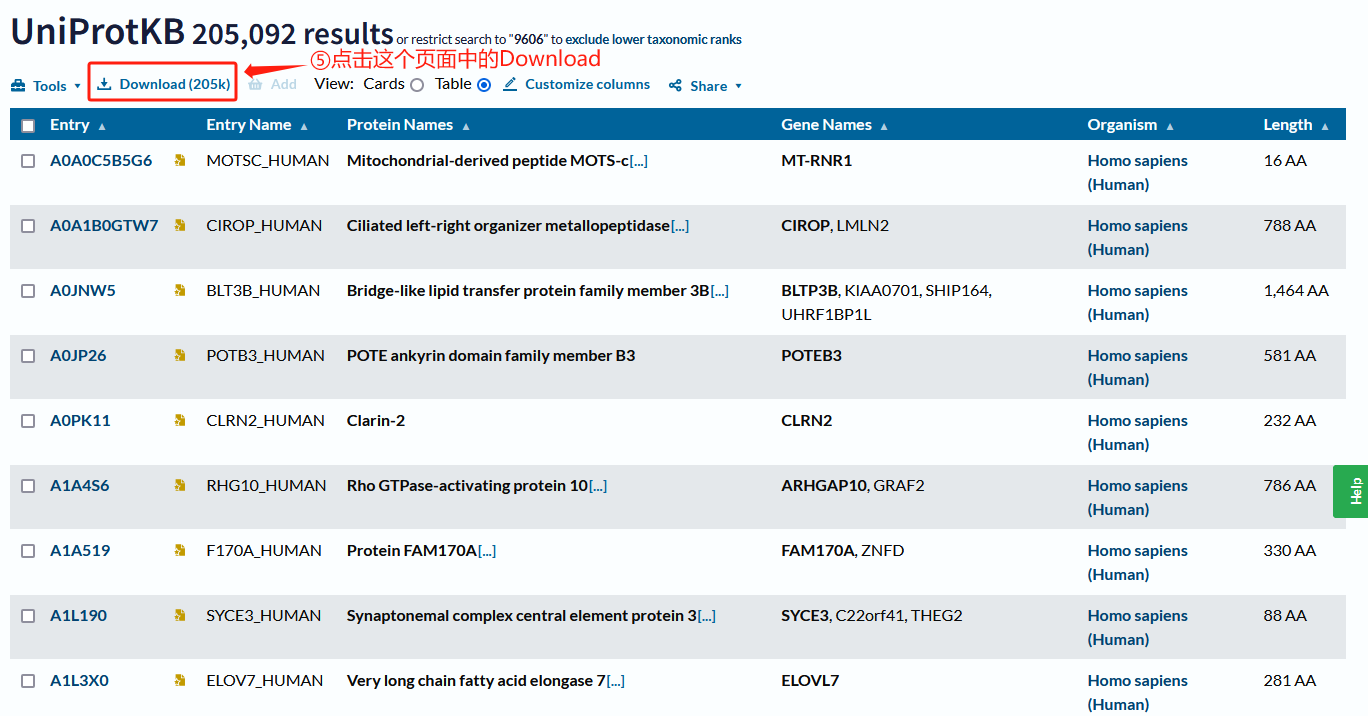
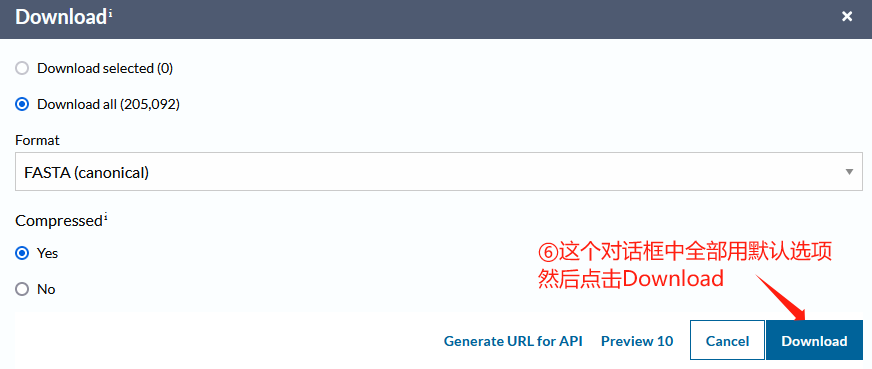
3.4 定量
假设上一步下载好的文件为 uniprotkb_HUMAN.fasta.gz 。我们将其和待处理的psm文件放在一起(见本文4.2小节。文件路径为 crux-output/psm-convert.txt ),然后先解压,再运行crux:
1 | # 解压缩。crux似乎不支持读取经过压缩的fasta文件 |
这将会在 crux-output 目录下生成三个文件:
1 | spectral-counts.log.txt |
其中的 spectral-counts.target.txt 就是我们所需的质谱定量文件。