测序技术与多组学技术概论
这一部分内容来自第四次助教课的备课笔记与PPT。主要梳理了一代至三代测序技术的原理和应用范畴,以及基于高通量测序的组学技术。
一、测序技术的简要发展史及其分类
测序技术大致可以分为三代:
- 一代测序(基于电泳的测序方法,代表技术如sanger测序)
- 二代测序(基于PCR的短读长测序,代表技术如illumina桥式PCR测序和华大基因DNA nanoball测序)
- 三代测序(单分子长读长测序,代表技术如PacBio SMRT测序和Oxford Nanopore测序)

测序技术发展很快,某种程度上已经超越了摩尔定律 (Moore’s law)——「在2007年,一次单遍测序最多可以产生的数据量约为1Gb。到2011年,这一 数值已经达到了1Tb,在四年的时间里约增长了1000倍。正是源于NGS这种可以快速产生大 量测序数据的能力,使得研究人员能够在几个小时或几天的时间里快速地将研究思路转换成完 整的数据集合。目前,研究人员可以在一次单遍测序中完成超过五个人类基因组的测序,产生 测序数据的时间约为一周,用在每个基因组上试剂的花费不超过5000美金。」
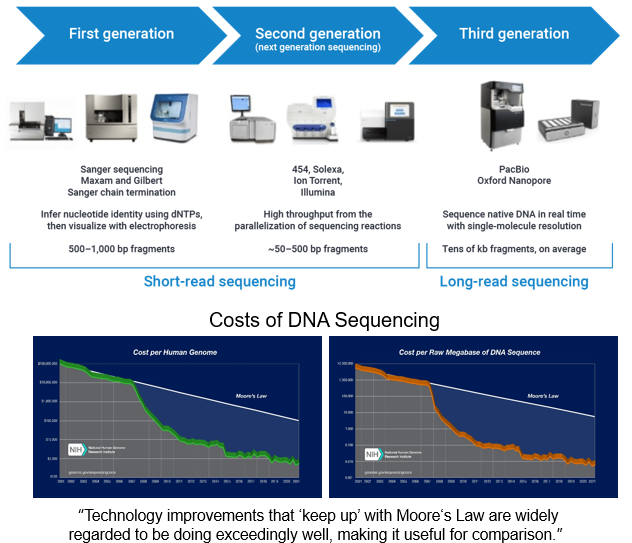
需要注意的是,二代测序和三代测序的划分其实有点模糊。一般来说,二代测序的特点是需要PCR扩增,且读长较短;三代测序的特点是单分子测序(不需要PCR扩增)且读长较长。按照这样的划分方法,Roche-454、SOLID、illumina solexa、DNBseq这些很明确属于二代测序,而PacBio、Nanopore很明确属于三代测序。
而Helicos测序(使用荧光标记的核苷酸与单分子模板杂交,并通过荧光信号识别碱基)、Ion torrent(检测DNA合成过程中释放的氢离子(H⁺)来确定核苷酸的序列)有点不好区分,它们既有二代测序的特点(读长较短),也有三代测序的特点(单分子测序,不需要PCR扩增)。
另一种划分方法是直接把上述这些测序方法归类为Next Generation Sequencing技术,不加以区分。
二、测序原理详解
第一代测序技术
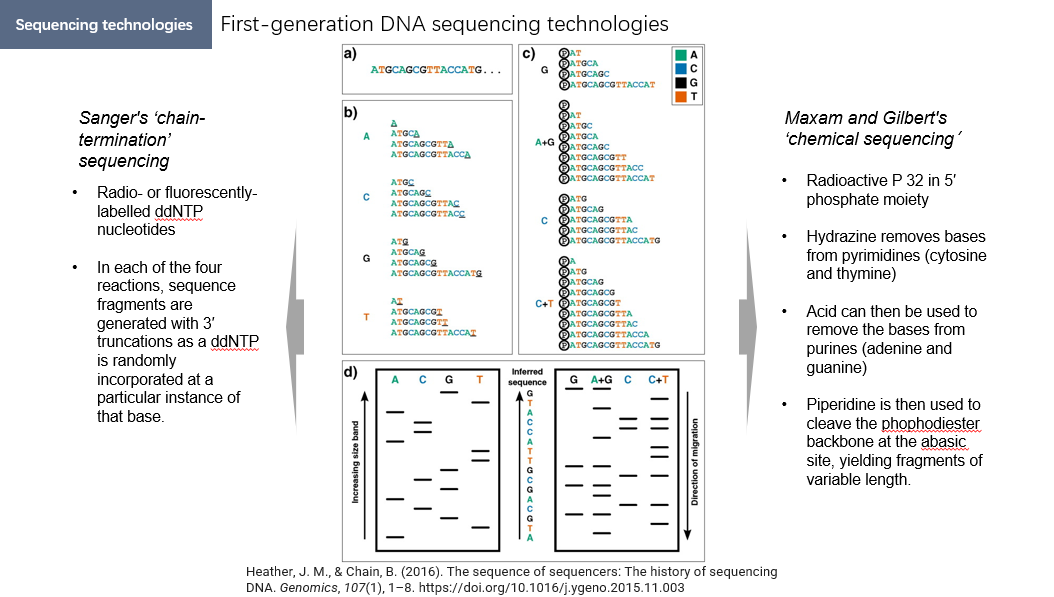
1977年Sanger对DNA的酶法测序技术作了改进,提出了 “双脱氧核苷酸末端终止法” 。其反应体系也包含单链模板、引物、4种dNTP和DNA聚合酶。共分4组,每组按一定比例加入一种2,3双脱氧核苷三磷酸,它能随机掺入合成的DNA链,一旦掺入DNA合成即终止,于是各种不同大小片段的末端核苷酸必定为该种核苷酸。经变性胶电泳,可从自显影图谱上直接读出DNA序列(上图左图)。 终止法测序极为方便。目前基于这种原理的测序仪一般将凝胶电泳改为毛细管电泳,从而进一步降低成本、提高测序效率。
化学法测序由Maxam和Gilbert于1977年所发明。其基本原理是用特异的化学试剂作用于DNA分 子中不同碱基,然后用哌啶切断反应碱基的多核苷酸链。用4组不同的特异反应,就可以使末端标记的 DNA分子切成不同长度的片段,其末端都是该特异的碱基。经变性胶电泳和放射自显影得到测序图谱。 4组特异的反应如下:
- (1)G反应,用硫酸二甲酯(dimethyl sulfate,DMS)使鸟嘌呤上的N原子甲基化,加热引起甲基化鸟 嘌呤脱落.多核苷酸链可在该处断裂。
- (2)G+A反应用,甲酸使A和G嘌呤环上的N原子质子化,从而使其糖苷键变得不稳定,再用哌啶 使键断裂。
- (3)T+C反应,用肼使T和C的嘧啶环断裂,再用哌啶除去碱基。
- (4)C反应,当有盐存在时,只有C与肼反应,并被哌啶除去。哌啶促使修饰碱基脱落,并使去掉碱基的磷酸二酯键断裂,上述样品分别进行聚丙烯酰胺变性胶电泳,放射自显影,此过程与酶法测序相同。
第二代测序技术
二代测序的代表技术有罗氏454测序(油包水PCR)、illumina测序(桥式PCR)、华大基因DNBseq(DNA纳米球PCR)等。其核心原理都是使用PCR对待测文库进行扩增,然后使用传感器检测DNA链延长过程中释放出的荧光信号,基于荧光信号对DNA序列进行还原。
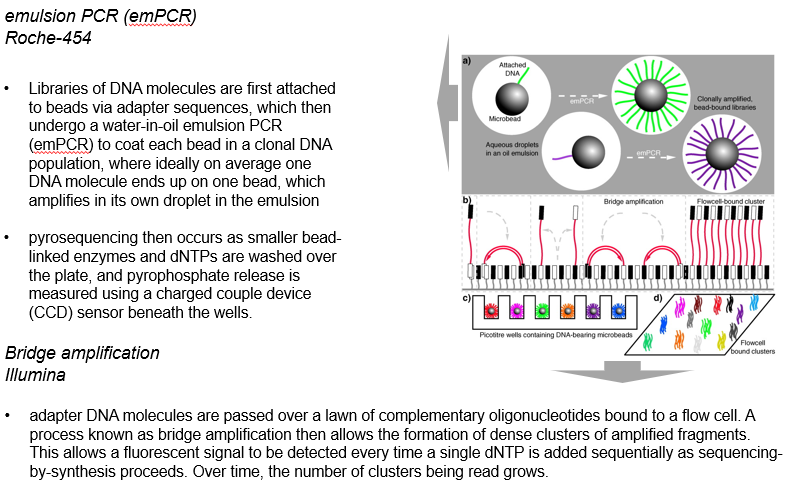

以illumina桥式PCR测序为例。桥式PCR是一种在固相支持物(玻片或芯片)上扩增DNA片段的技术,依靠引物固定于芯片表面,通过多轮PCR循环实现目标DNA的扩增,最终生成密集的DNA簇(cluster),为后续的测序提供足够的信号。
- 首先,待测DNA会被片段化为200-500bp左右的片段,并与接头序列连接进行PCR扩增。
- 随后,这些DNA会被装载到flow cell的反应体系中:正向引物和反向引物都被通过一个柔性接头(flexible linker)固定在固相载体(solid substrate)上,待测的DNA会与这些引物互补配对从而装载到反应体系。
- 之后,经过桥式PCR反应,所有的模板扩增产物就都被固定到了芯片上固定的位置。值得注意的是,Illumina测序仪使用的桥式PCR与传统的桥式PCR有所不同,它会交替使用Bst聚合酶进行延伸反应以及使用甲酰胺(formamide)进行变性反应。这样,经过桥式PCR扩增之后,也会在固相载体上形成一个个的模板“克隆”。一块芯片的8条独立“泳道”上每一条泳道都可以容纳数百万的模板“克隆”,这样一次就可以同时对8个不同的文库进行测序。
- illumina采取的是一种边合成边测序(Sequencing by Synthesis, SBS)的策略,即测序阶段使用带荧光标记的可逆末端终止核苷酸(reversible terminators)作为反应底物,每次加入一个核苷酸,通过荧光信号检测DNA链的延伸情况,并将其转化为碱基序列。
此处有两个思考题:
- Why Bridge amplification?
- Limitations of NGS?
答案:
- easily detect it with a microscopy system; makes the signal more robust
- As going through more cycles of chemistry the signal in the different molecules in a cluster get more out of sync with each other (Illumina call this a loss of phasing ). You can correct some of this computationally, but eventually the signal just breaks down so that your signal: noise gets so bad that you can’t reliably call bases any more.
第三代测序技术
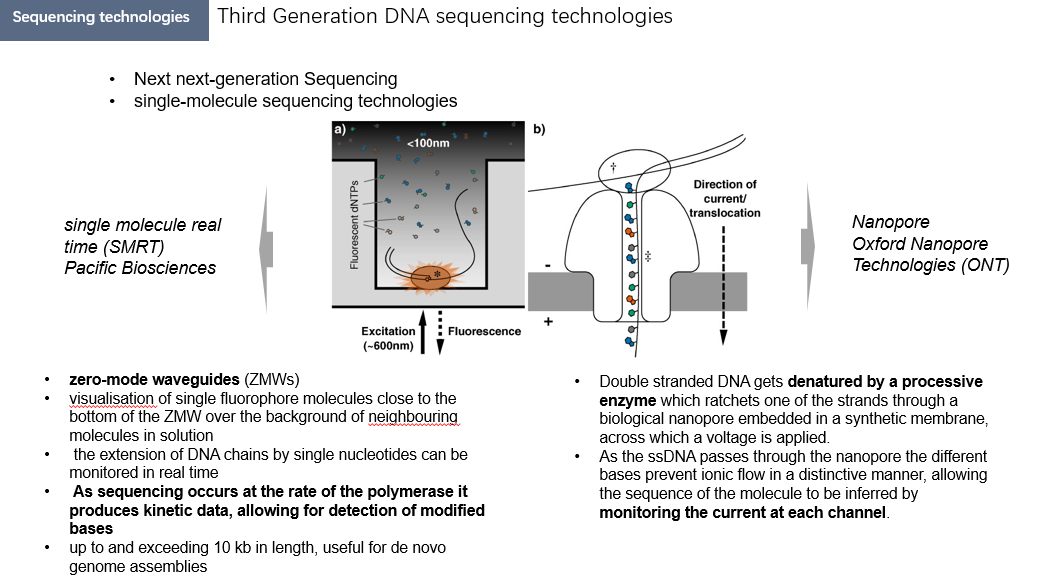
三代测序技术目前常用的有两种,分别是基于荧光信号的PacBio SMRT测序和基于电信号的Oxford Nanopore测序。
PacBio SMRT测序技术的原理基于单分子实时测序(Single-Molecule Real-Time Sequencing),通过在纳米级孔洞内捕获DNA分子并实时记录酶的合成活动实现测序。在这一技术中,DNA分子被环化形成“发夹结构”(SMRTbell),并通过特定的DNA聚合酶固定在零模波导孔(ZMW)内。ZMW是一个极小的光学孔洞,能将激光聚焦到单分子水平,仅照亮固定区域的DNA分子。聚合酶在ZMW中通过添加带荧光标记的核苷酸对DNA进行复制,每次核苷酸的加入会释放特定波长的荧光信号。荧光信号由检测器实时捕获并记录,而荧光基团在核苷酸加入后被及时切除,不会干扰后续反应。通过分析荧光信号的颜色和持续时间,可以直接解读DNA的碱基序列。由于不需要扩增,SMRT测序避免了PCR相关的偏差,能直接读取数万碱基的长DNA片段,适合分析复杂基因组和重复序列。
Oxford Nanopore测序技术依赖于一种基于电信号的原理,通过监测单链DNA穿过纳米孔时产生的电流变化来解读序列信息。在这一技术中,DNA双链被解链为单链并通过特定的接头加载到纳米孔装置中。纳米孔由生物分子(如α-溶血素蛋白)或固态材料制成,嵌入到电流流通的膜中。当DNA单链在电场的驱动下通过纳米孔时,不同碱基会对孔内的离子流产生特定的阻碍作用,导致电流产生可检测的变化。通过实时分析电流信号的特征,系统可以推断出DNA的碱基序列。Oxford Nanopore测序技术具有便携性强、读长可达数十万碱基的特点,同时还可以直接测定DNA的修饰信息(如甲基化)。这种技术在实时测序和远程应用(如野外检测和传染病监控)中具有显著优势。
对比(优势与不足,以及各自的使用场景)
| 对比项目 | 一代测序(Sanger测序) | 二代测序(基于PCR的短读长测序) | 三代测序(单分子长读长测序) |
|---|---|---|---|
| 测序原理 | 基于双脱氧核苷酸链终止法,通过毛细管电泳分离扩增片段并读取荧光信号。 | 基于PCR扩增的短片段库构建,利用可逆末端终止技术(Illumina)或焦磷酸测序技术(Roche 454)。 | 直接检测单分子DNA的序列变化,PacBio基于荧光信号,Nanopore基于电流信号。 |
| 测序成本 | 每碱基成本较高,单次运行成本适中(适合小规模样本)。 | 每碱基成本最低,适合大规模高通量测序,但设备成本较高。 | 每碱基成本较高,但单次运行生成数据量大,适合分析复杂样本。 |
| 测序读长 | 中等(500-1000 bp)。 | 短(通常50-300 bp)。 | 长(可达10-100 kb,甚至更长)。 |
| 错误率 | 低(~0.1%)。是测序技术的金标准。 | 较低(~0.1%-1%),依赖于高覆盖率纠正。 | 高(10%-15%,通过深度覆盖和后处理可降低到<1%)。 |
| 通量 | 低(适合单个或少量样本)。 | 高(适合大规模基因组或转录组测序)。 | 高,但单次运行时间较长,适合全基因组和表观遗传学分析。 |
| 应用领域 | 小规模基因验证、单基因突变分析、特定片段测序。 | 全基因组测序、转录组分析、突变检测、微生物群落分析。 | 复杂基因组分析(如重复序列、结构变异分析)。 |
| 时间效率 | 较慢,需进行片段分离和单一扩增反应。 | 快速,高通量能一次完成大量片段的测序。 | 适中,实时测序但分析复杂且需纠正高错误率。 |
| 适用样本规模 | 小样本规模(如一个基因或小型病毒基因组)。 | 中到大规模样本(如人类基因组、癌症研究中的多样本)。 | 大规模样本、复杂样本(如古DNA、大型植物或动物基因组)。 |
三、文库制备技术与下游分析pipeline
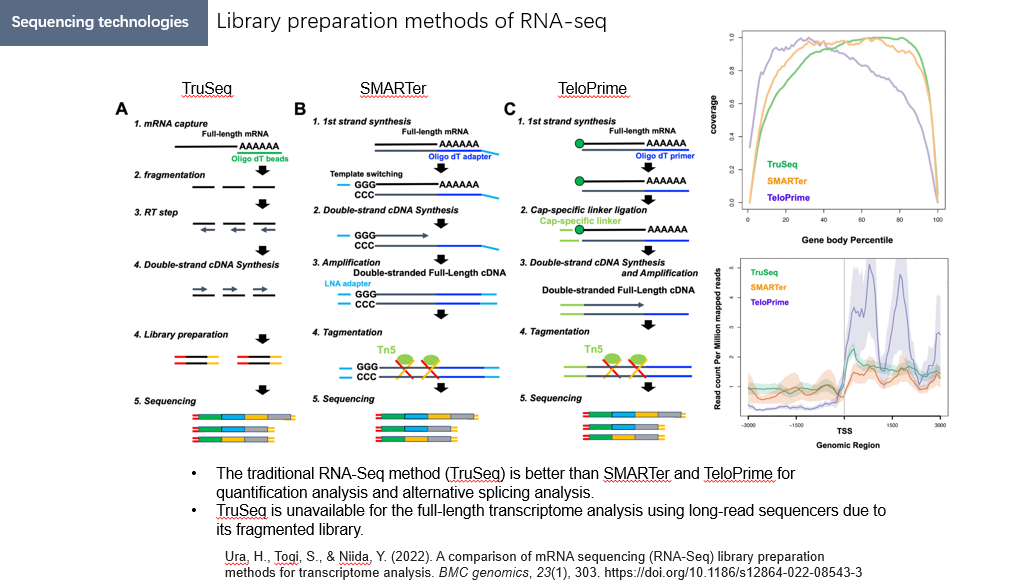
以RNA-seq为例。我们要测的是细胞质中的mRNA,但是上述测序技术的样品是DNA,因此需要经过一些文库制备的过程。
这一页PPT列出了三种不同的文库制备技术:传统RNA-seq文库制备(TruSeq)、针对PacBio SMRT长读长测序仪的文库制备(SMARTer),以及主要用于分析TSS位点的TeloPrime测序。
TruSeq
- 原理:采用传统的RNA-Seq方法,首先通过oligo dT珠捕获mRNA后,随机剪切成片段,并进行反转录生成cDNA,再通过随机引物合成双链cDNA。
- 特点:mRNA片段化处理有助于均匀覆盖基因的全长。
- 应用:适用于短读长测序(short-read sequencing)的转录组定量分析和剪接事件检测。
- 优势:对转录本和剪接事件的检测能力较强,适合需要均匀覆盖基因全长的分析。
SMARTer
- 原理:利用MMLV(Moloney Murine Leukemia Virus)逆转录酶的模板转换(template switching)特性,生成全长双链cDNA。
- 特点:保留了完整的转录本,但对于长转录本表现出一定的扩增偏差。
- 应用:适用于长读长测序(long-read sequencing),如PacBio或Oxford Nanopore平台,用于全长mRNA结构的解析。
- 优势:在非特异性基因组DNA扩增风险可控的情况下,可进行详细的转录本结构分析。
TeloPrime
- 原理:基于帽结构特异性接头连接(cap-specific linker ligation)技术,从完整的5′帽状mRNA分子生成全长双链cDNA。
- 特点:对转录起始位点(TSS)的覆盖率较高,但在基因体其他区域的覆盖较差。
- 应用:适合分析转录起始位点(TSS)特异性数据的研究。
- 优势:在研究转录起始区域时表现较佳,但对基因其他区域覆盖不够均匀。
关于多组学分析的pipeline,下面两页PPT中推荐了两篇综述,感兴趣的读者可以自行下载和阅读。
Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., Szcześniak, M. W., Gaffney, D. J., Elo, L. L., Zhang, X., & Mortazavi, A. (2016). A survey of best practices for RNA-seq data analysis. Genome biology, 17, 13. https://doi.org/10.1186/s13059-016-0881-8
Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., & Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome biology, 21(1), 30. https://doi.org/10.1186/s13059-020-1935-5
四、基于高通量测序的组学技术
下面列举的这些技术,虽然名字都叫”XXseq”,但严格意义上来说他们并不是一种新的测序技术(基本上都属于二代测序的范畴),而是在测序技术的基础上检测细胞中的不同核酸类型,以服务于各种组学研究。因此,最好的称谓还是“基于高通量测序的组学技术”。
ATAC-seq
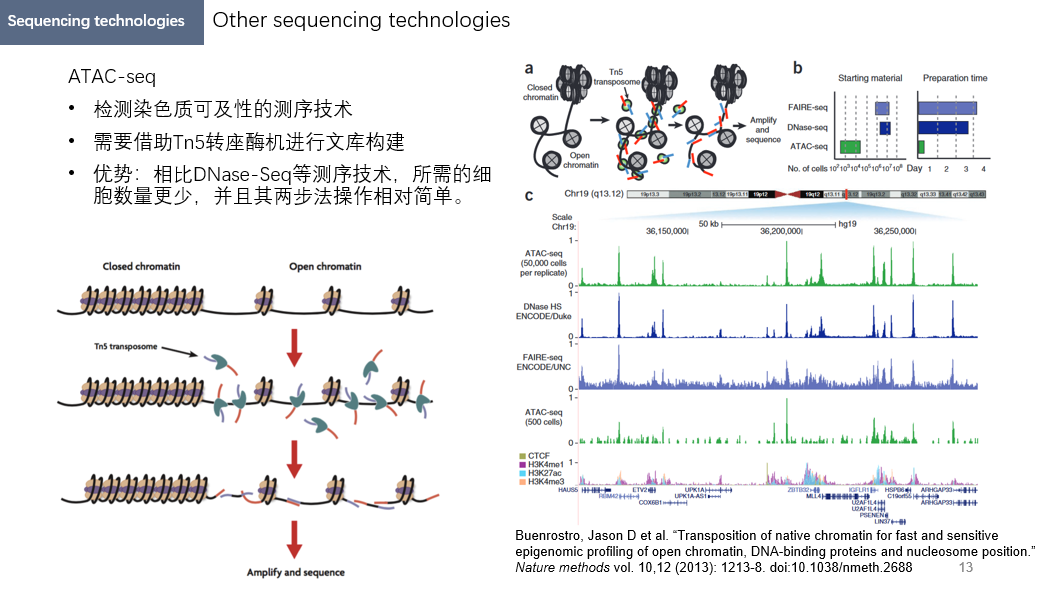
开放染色质区测序(ATAC-seq)是一种检测全基因组染色质开放区域的组学技术,其原理是使用Tn5转座酶在染色质开放区域插入接头,直接打断和标记DNA。沉默的染色质区域一般会有大量蛋白结合,导致Tn5转座酶难以结合,而开放染色质区则相对容易结合,基于这一点即可分辨开放染色质区的位置。
ChIP-seq
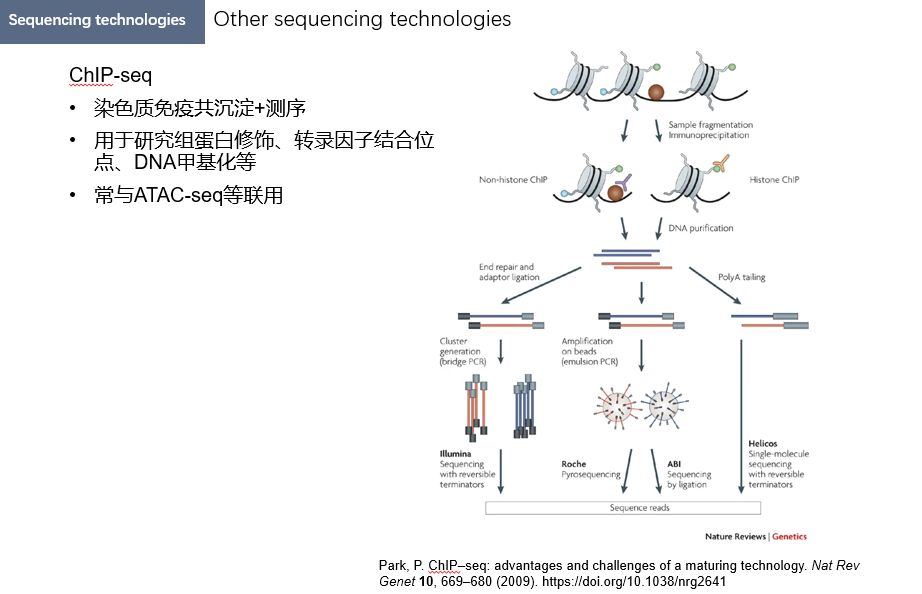
染色质免疫共沉淀测序(ChIP-seq)是一种结合了染色质免疫共沉淀(ChIP)和二代测序的组学技术。使用特异性抗体富集特定蛋白结合的DNA(如转录因子、组蛋白修饰),提取富集的DNA片段并测序,即可检测出这些特定蛋白结合的DNA的位置。用于研究组蛋白修饰、转录因子结合位点、DNA甲基化等科学问题,常与ATAC-seq、DNase-seq、MNase-seq等技术联用。
Ribo-seq
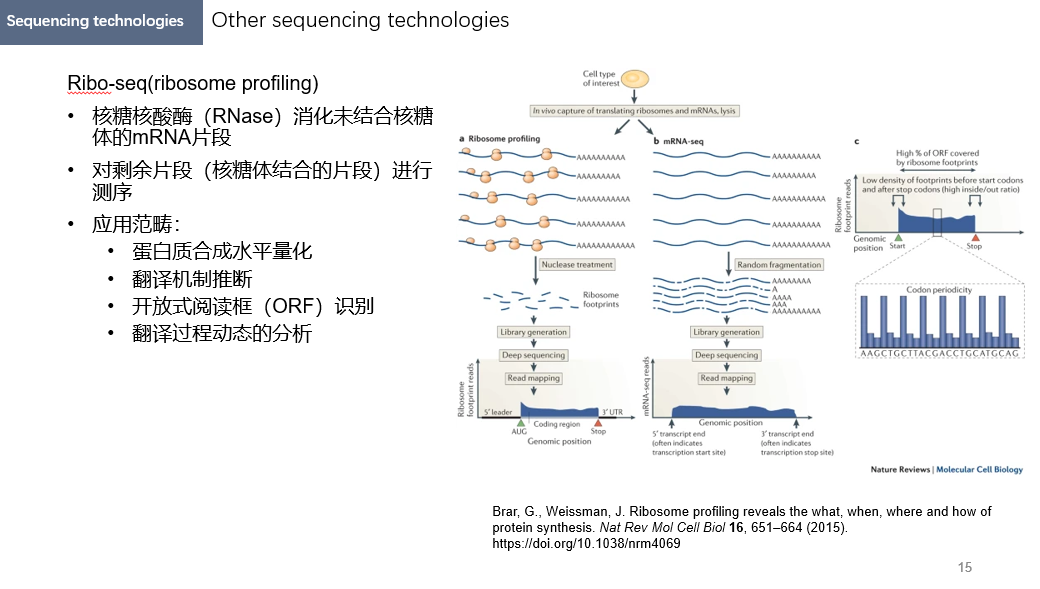
核糖体测序(Ribo-seq)也叫核糖体分析(ribosome profiling),使用环己酰亚胺(CHX)等翻译抑制剂稳定核糖体在mRNA上的结合,并消化未结合核糖体的RNA片段,对剩余mRNA片段进行富集和测序,从而可以确定核糖体的结合位点。常用于分析翻译中的活跃mRNA片段(翻译组学)、鉴定起始密码子/翻译暂停位点/非经典翻译事件、研究翻译效率、翻译调控机制以及蛋白质生成速率、发现非编码RNA的潜在翻译功能。
HiC与三维基因组
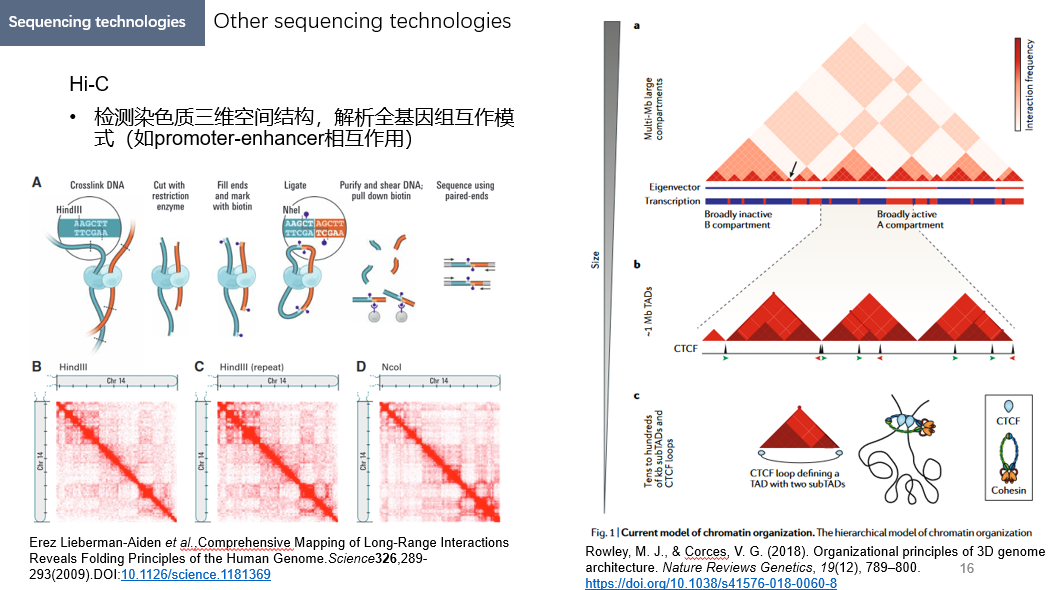
这是一种用于研究染色质高级结构的组学技术。
关于染色质高级结构,可以参考综述 “Organizational principles of 3D genome architecture” ,简单来说就是染色质内部存在不同区室(compartment,分辨率在数个MB左右)的划分,这些区室内部有很强的互作,而区室之间则互作很少。区室可以进一步划分为拓扑关联域(topologically associating domains,TAD,分辨率大约1MB),而TAD还可以进一步划分为多个由CTCF蛋白结合位点标记的CTCF loop。这些不同层次上的高级结构,是启动子、增强子、绝缘子等调控元件发挥功能的关键。
对于HiC来说,其原理如下:空间上有互作的DNA,其距离会很近。因此,提取细胞核,使用甲醛固定染色质(让正在互作的DNA-蛋白质和DNA-DNA交联在一起),随后用限制性内切酶消化DNA并用带接头的核苷酸填充,之后使DNA环化,这样空间上互作的DNA就会被保留在同一条reads内。对其进行测序,即可分析染色质三维结构(如环状结构、拓扑关联结构域TAD),并帮助我们理解基因调控元件的空间作用、细胞分化和疾病中染色质结构变化。
其他
还有许多基于高通量测序的组学技术,例如 RIP-seq, PRO-seq, CLIP-seq, meDIP-seq 等,它们在一些特定研究领域发挥着非常重要的功能。
限于篇幅,此处不详细讲,并作为课后作业供同学们自行查询和了解。感兴趣的读者朋友们也可以自行查找资料或者借助AI工具了解这些技术。