文献阅读笔记:Universal Patterns of Selection in Cancer and Somatic Tissues
近期阅读了一篇研究癌症中的somatic mutation的文章,下面是阅读过程中的一些笔记。
这篇文章于2017年发表在Cell期刊上,下面是citation信息:
Martincorena, Iñigo et al. “Universal Patterns of Selection in Cancer and Somatic Tissues.” Cell vol. 171,5 (2017): 1029-1041.e21. doi:10.1016/j.cell.2017.09.042
摘要编译
癌症是体细胞突变(somatic mutation)和克隆选择(clonal selection)的结果,但目前缺乏对癌症进化过程的选择压力进行定量的方法。这篇文章将分子进化领域的方法(即dN/dS)应用在了癌症研究当中,分析了29种癌症类型共7664种肿瘤。
与物种进化不同,在癌症的发展过程中,正选择事件多于负选择事件。平均而言,每个肿瘤中负选择事件导致的碱基替换的丢失不到1个(肿瘤中的这些碱基替换基本上是有害的,如果负选择正常,那么应该有更多的碱基替换被清楚,而不是保留),除了必需基因的纯合丢失外,几乎不存在纯化选择。
上述特性也允许我们对所有编码区的驱动突变(driver coding mutations)进行全基因组计数,包括那些我们未知的癌症基因。平均而言,由于正选择,一个肿瘤平均携带约4个驱动突变(从甲状腺癌和睾丸癌的不到1个,到子宫内膜癌和结直肠癌的多于10个)。一半的驱动程序替换发生在我们未知的癌症基因中。
随着突变负担的增加,驱动突变的数量也会增加,但二者的关系不是线性的。这篇文章系统地对癌症基因进行了分类,并表明基因在驱动突变(driver mutation)和乘客突变(passenger mutation)的比例上有很大差异。
作者简介
本文的主要作者均来自sanger实验室。第一作者Inigo Martincorena的主要研究方向是somatic mutation在cancer、aging中的作用。他开发了dndscv这一工具(即本文),将dnds这一分子进化领域的计算工具应用在了体细胞突变的研究当中。通讯作者Peter Campbell的主要兴趣点包括新癌症基因的发现、肿瘤的体细胞突变过程鉴定、癌症进化的模式等,他还参与了sangger实验室的Cancer aging and somatic mutation项目,这一项目有许多著名的成果,如记录肿瘤突变特征的COSMIC数据库,以及cancer genome browser等。
研究背景
1. 体细胞突变的研究现状
癌症是体细胞进化的最终产物,在体细胞进化中,单个克隆谱系获得了驱动突变的补充,使细胞能够逃避细胞增殖的正常限制,入侵组织,并扩散到其他器官。
然而有两个 科学问题 仍未得到解答:
- The number of mutations required to drive a cancer(驱动癌症所需的突变数量)
- Whether this number varies extensively across tumor types or with different mutation rates(突变数量是否因肿瘤类型或不同的突变率而广泛变化)
解决这个问题的一种方法是使用年龄变化曲线来估计癌症发展所需的减缓速率步骤的数量,隐含地假设减缓速率步骤和驱动突变之间的一一对应关系。然而,并非所有驱动突变都需要限速(Yates等人,2015),也不需要每个限速事件都是驱动突变(Martincorena和Campbell,2015)。
第二种方法只是计算已知癌症基因中发生的突变,但当前我们所知道的癌基因列表并不完整(还有许多癌基因有待发现),因此这种方法的统计结果也不准确。因此,亟需新方法的开发。
2. 一种检测选择压力的方法:Ka/Ks(也称为dN/dS)
在遗传学中,Ka/Ks表示的是两个蛋白编码基因的异义替换率(Ka)和同义替换率(Ks)之间的比例,这个比例可以判断是否有选择压力作用于这个蛋白质编码基因(Hurst LD, 2002)。
Ka和Ks的计算公式:
- Ka=发生异义替换的SNP数/异义替换位点数
- Ks=发生同义替换的SNP数/同义替换位点数
其中同义替换位点数就是不会造成氨基酸变化的位点数的总和,比如编码丝氨酸(serine,Ser)的密码子TCT的第三位碱基(这一位点的三种替换结果TCA、TCG、TCC都编码丝氨酸,因此这个位点算1个同义替换位点)。而异义替换位点数就是会造成氨基酸变化的位点数的总和,比如编码丝氨酸的密码子的第一二位碱基。另外,计算Ka/Ks时,不考虑起始密码子(start codon)和终止密码子(stop codon)。
一般情况下,在某个个体中偶然发生的一个碱基替换(突变),如果没有额外的好处或者坏处的话,慢慢地也就消失了。但是自然选择中会有很多巧合,某些突变就是很幸运地被保留了下来,并且被固定了(突变频率由极小变为100%)。一个这样的突变在一个二倍体种群中被固定的可能性为1/2N,其中N是种群大小。
对于一个没有受到自然选择压力的基因来说,Ka近似等于Ks,也就是说Ka/Ks≈1。但有些情况下,异义突变(非同义突变,nonsynonymous mutation)会带来更加有益或更加有害的性状,从而受到自然选择。当正选择作用在一段序列上时,异义突变的结果倾向于被保留,也就是Ka/Ks>>1;而当负选择(纯化选择)作用在一段序列上时,异义突变的结果倾向于被清楚,也就是Ka/Ks<<1。

人们已经提出了许多Ka/Ks算法,这些方法按照原理大致可以分为计数法和似然法,前者主要通过统计序列比对中的同义替换和异义替换数对Ka/Ks进行估算,代表方法如NG86法(Nei, 1983)和LWL85法(Li WH, 1985)等;后者则通过碱基替换率的似然值函数对Ka/Ks进行估算,代表方法有GY94(Yang Z, 2000)和PAML(Yang Z, 2007)等。一般来说,计数法方便快捷,适合快速计算Ka/Ks;而似然法虽然计算量更大,但也估计得更精准。

3. 本文对Ka/Ks计算框架的改进措施
前面提到的Ka/Ks的计算框架主要用于物种层面上的进化分析。相比之下,体细胞携带的碱基差异数更少,且更容易受到基因组上下文(context)的影响,因此方法需要改进。
本文作者使用泊松分布框架对突变信息进行建模。使用泊松分布的原因包括(1)泊松分布适合稀疏且独立的事件的建模,就像somatic mutation这种(2)泊松分布参数简单易于推导(3)好做似然比检验(4)稳健性好。
第一点改进:在检测时考虑突变上下文信息。体现在似然模型构建的过程中,就是使用一个多达192个参数的突变参数,用于分辨不同的突变类型(按照作者的分法,就是TAT>TTT用一个参数统计,TAC>TTC也要用一个参数统计,这两种本质是同一个碱基发生的突变,但是相邻核苷酸不同,因此需要分开算)。

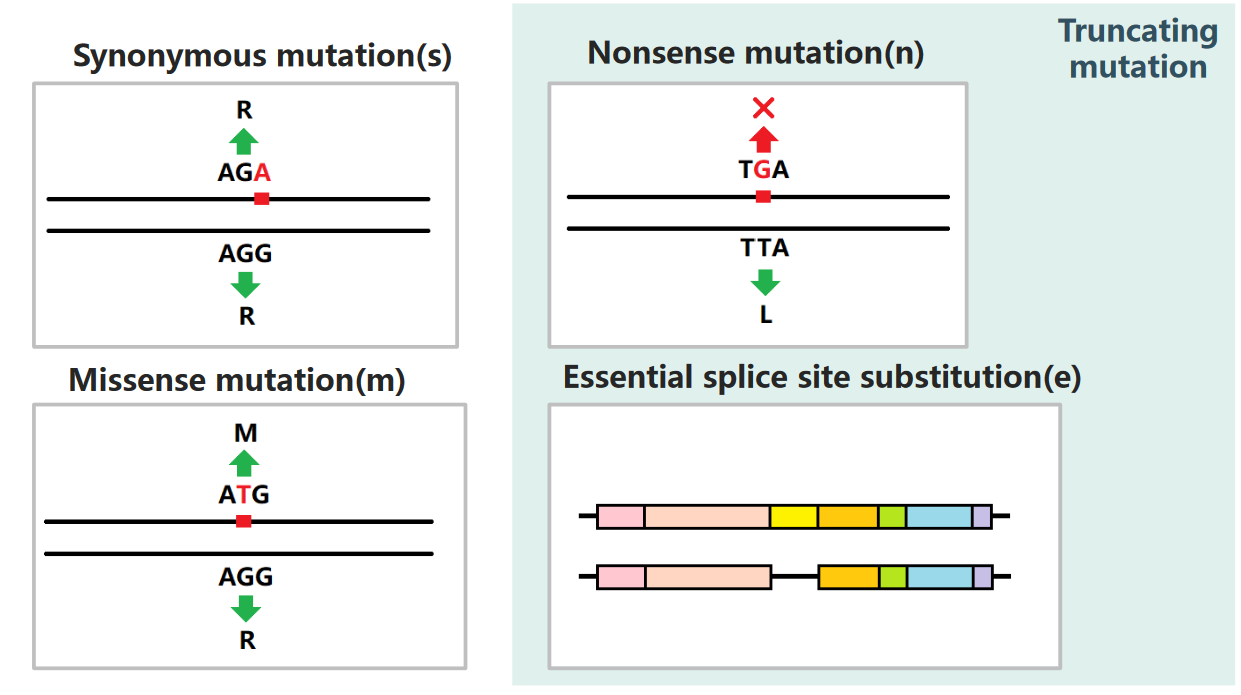
第二点改进:传统的Ka/Ks计算框架只考虑非同义突变(即missense mutation),但是这里作者通过增加模型参数,把无义突变(nonsense mutation)和关键剪接位点的突变也考虑了进去。
第三点改进:采用更加严格的突变筛选流程,以避免种系突变(germline mutation)造成的偏差。
- Extreme caution was exercised during variant calling
- to avoid biases emerging from germline variants
- because these have a much lower dN/dS ratio than somatic mutations.
第四点改进:考虑到体细胞突变可能受到基因组局部突变率的影响,作者使用一个负二项分布广义线性模型(glm.nb)对单个基因的背景突变率进行矫正。使用这一矫正的模型被作者称之为dNdScv,是本文提出的一个重要模型。
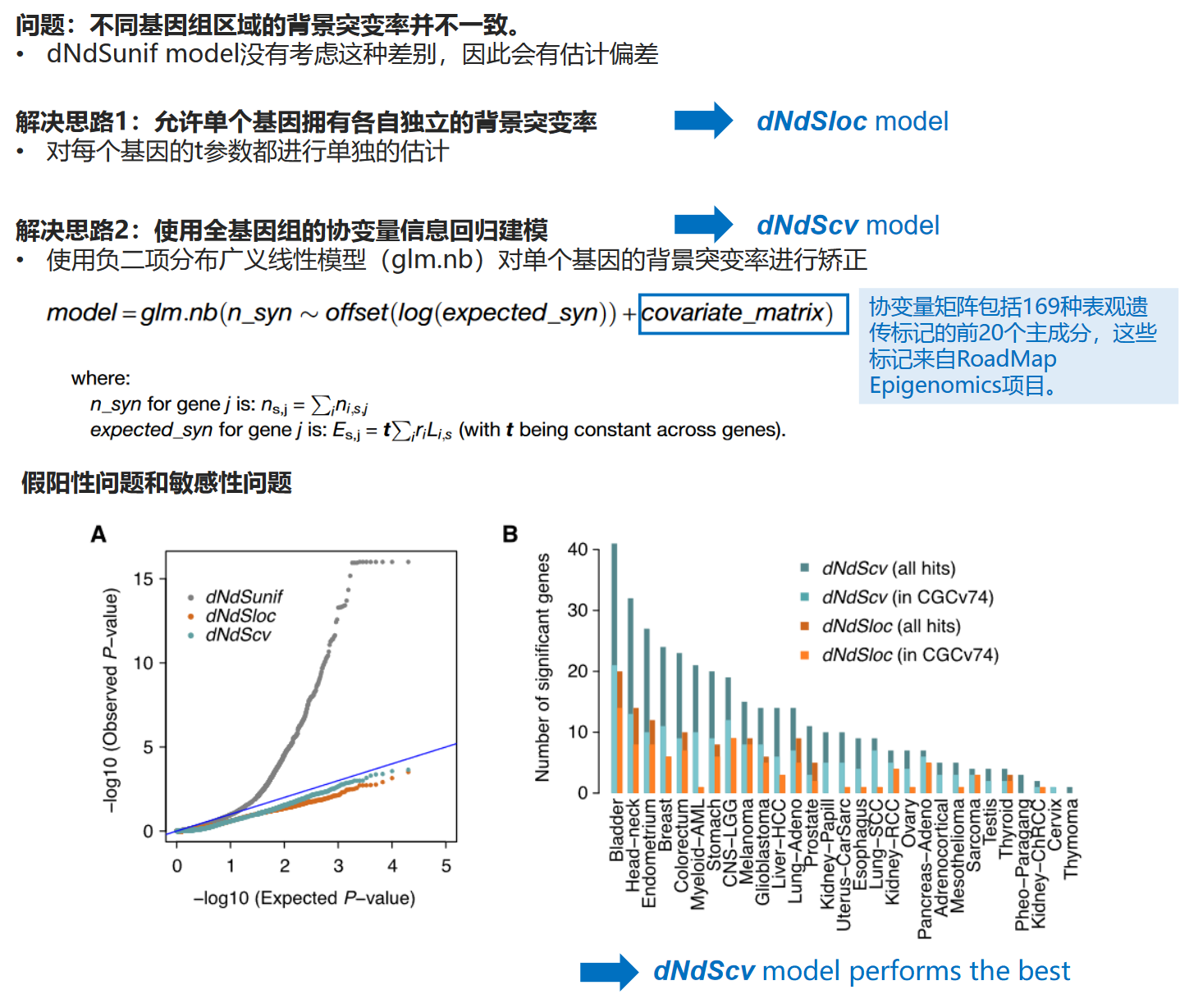
offset(log(expected_syn))代表基因暴露的对数,它反映了基因大小、序列特征等因素对突变率的影响。covariate_matrix包括169种表观遗传标记的前20个主成分,这些标记来自RoadMap Epigenomics项目。- 模型假设每个基因的同义突变数服从负二项分布。
- 负二项分布是一种Gamma-Poisson复合分布,其中Poisson分布的均值遵循Gamma分布。
n_syn服从参数为λ的泊松分布,而λ又服从参数为a和b的Gamma分布。
上述负二项分布广义线性模型(glm.nb)的一些实现细节:
n_syn对于基因j是所有样本中该基因的同义突变总数。expected_syn对于基因j是所有样本中该基因的预期同义突变数。- 通过使用协变量矩阵,模型能够考虑表观遗传学因素对背景突变率的影响,从而改进背景突变率模型。
- 模型输出估计的背景突变率以及模型的过分散参数。
- 过分散参数(q)反映了突变率在不同基因间的变异情况,从而便于对局部突变率进行矫正。
dNdSunif/dNdSloc/dNdScv三种方法的比较:
- A图:ICGC数据库中107个随机melanoma数据集的统计结果的QQ-plot。注意,dNdSunif的中部和尾部过度上扬,表明假阳性过于严重,因此被放弃。
- B图:敏感性分析,对29个TCGA数据集的统计结果。dNdSloc的敏感性不足,因此被放弃,后续主要使用dNdScv方法去做分析。
一些重要的结果
1. 体细胞突变的选择压力模式
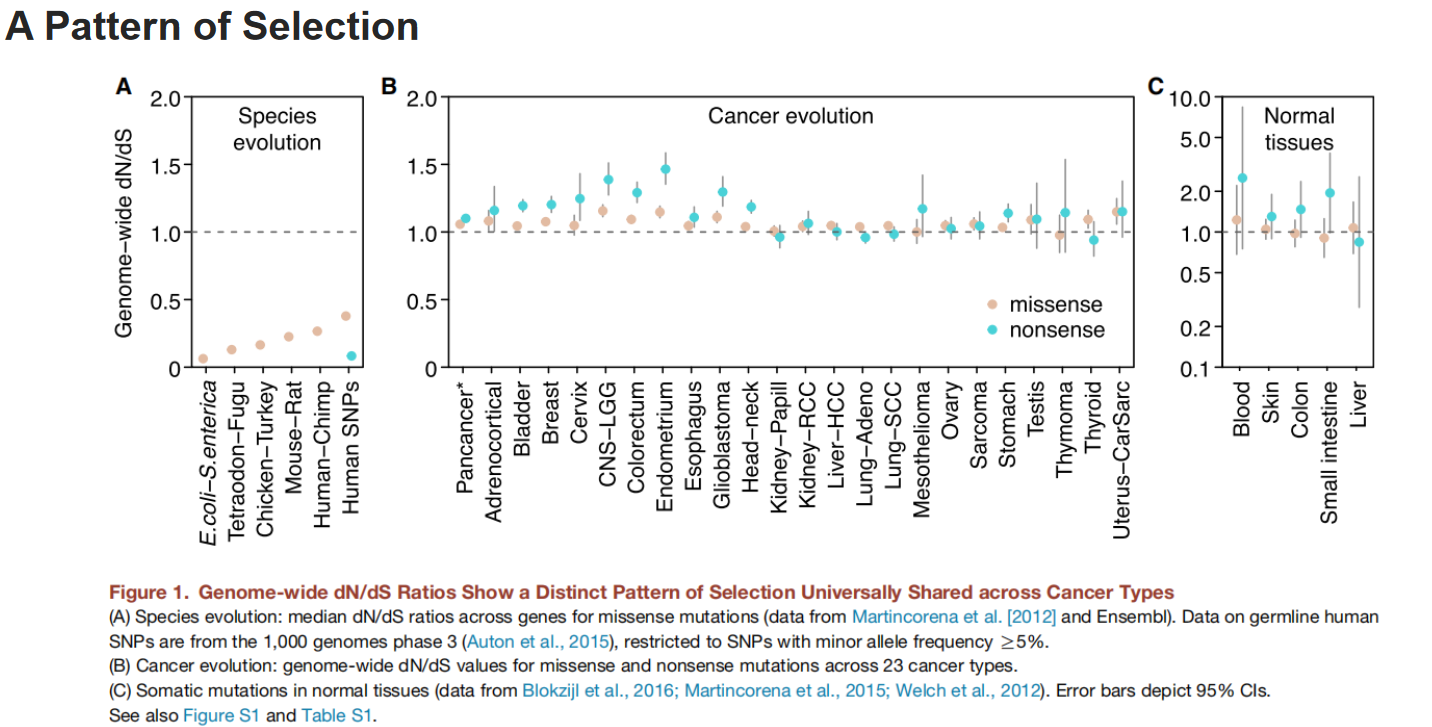
- A:跨物种的dN/dS比较。注意到,所有物种中的dN/dS普遍小于1,在常见的人类germline 多态性中,nonsense 突变的dN/dS比率也同 样很低(dN/dS≈0.08 )。
- B: 在癌症中,正选择压力下的突变比负选择的更多,但总体情况接近中性
- C: 但是前人研究也发现,一些正常人体组织中(如blood、skin、liver、colon、小肠),dN/dS也是略微大于1的【可能因为需要大量细胞分裂?】
2. 不同肿瘤中,受到正选择压力的基因,以及它们所受到的选择压力
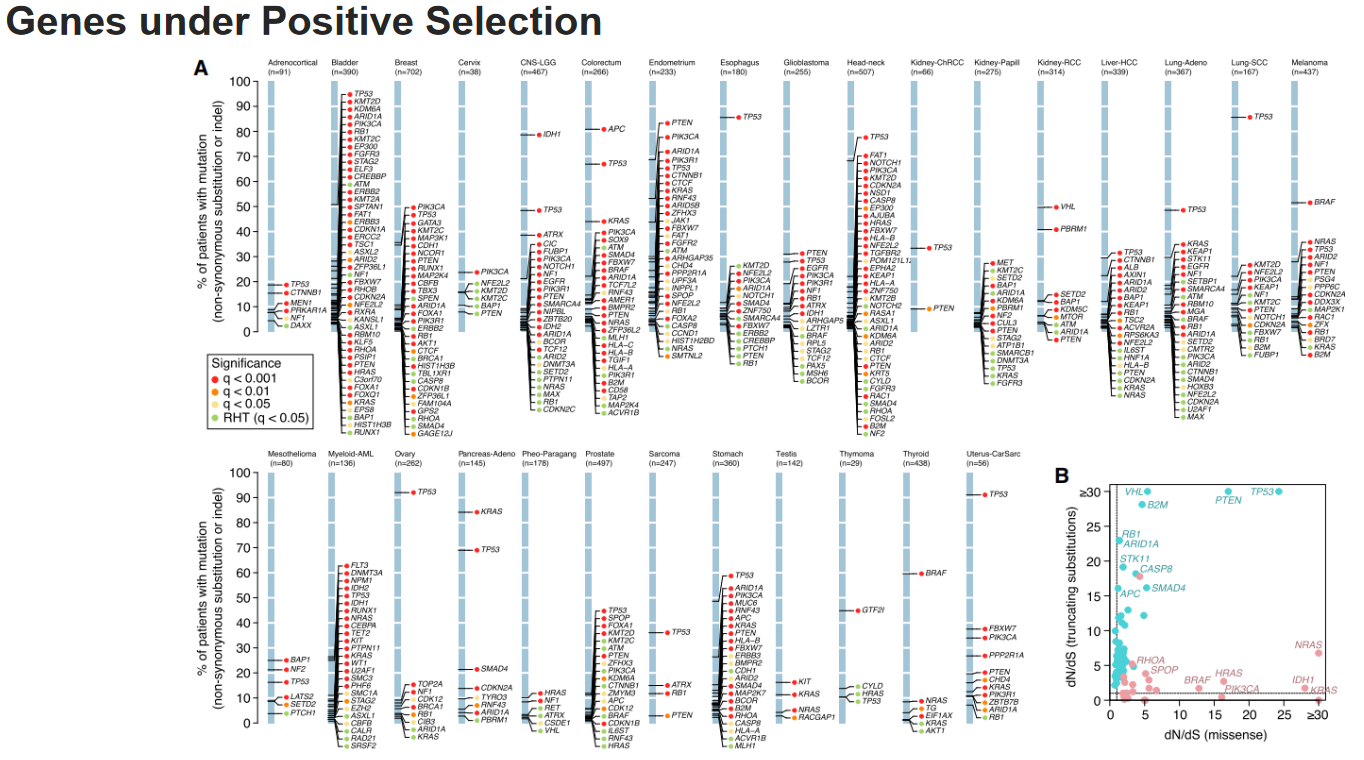
作者接下来去看了每个基因的dN/dS。注意,A图的竖线并非染色体,而是数据集中患者携带相关突变的百分比。一条竖线代表一种cancer。这里只展示dN/dS显著大于1的基因,可以注意到,一个基因被患者携带的比例越高(在竖线上的位置越靠上),则这个基因的显著性也相对越高。另外,TP53在多个cancer dataset中均有出现。
由于dndscv方法可以对missense和truncating突变分别做计算,作者也根据相关结果绘制了散点图,如B图。tranc dnds高而mis dnds低的多为抑癌基因(即,对truncating这类突变有正选择;图中绿点),反之则为原癌基因(对missense正选择,图中红点)
3. 在肿瘤中,负选择基本上不存在
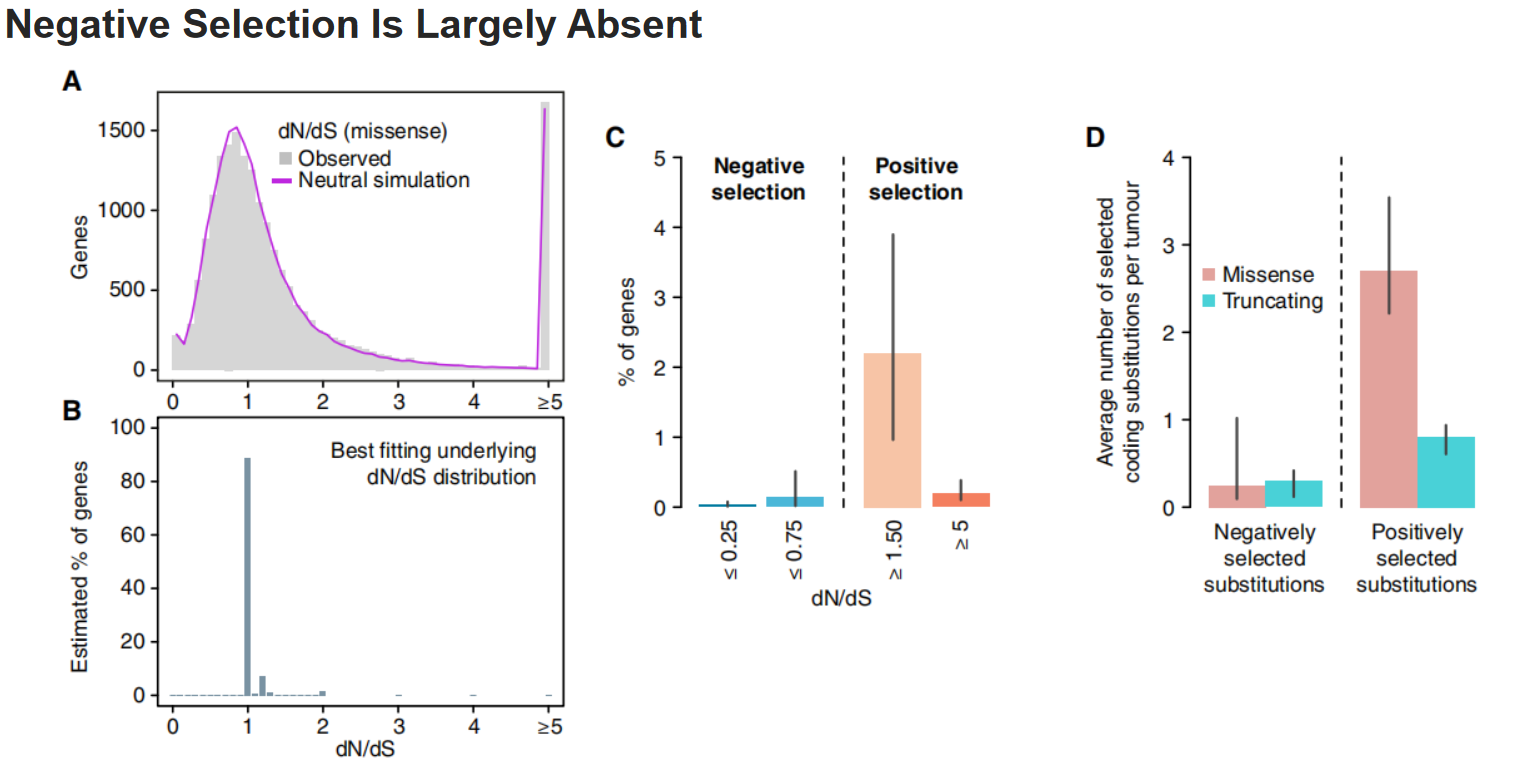
对体细胞突变的负选择早已被期待已久,但尚未在癌症基因组中得到可靠的记录。
- A: 统计了所有基因的missense mut dN/dS的分布情况,这一分布情况与中性条件下的模拟结果几乎一致。
- B: 作者认为A图的统计并不完美,因为每个基因有限的突变数量使单个基因的dN/dS值嘈杂。为了正式估计在负选择下的基因的比例,作者使用a binomial mixture model(二项分布混合模型)推断了不同dN/dS的基因的数量,推断结果如B所示,其中大部分基因Ka/Ks=1,少数基因Ka/Ks>1,极少数<1.。
- C: 是对B图的一个汇总统计。尽管正选择和负选择基因都很少,但是正选择基因依然稍多于负选择基因,表明在肿瘤细胞中对missense mut的负选择作用是缺失的。
- D: 前面统计的都是missense mut,因此作者也用同样的方法看了一下truncating mut(包括nonsense和splicing mut),发现负选择同样也是缺失的。需要注意,D图的纵坐标是每个肿瘤携带的突变数量,和C图略微不一样。
总之作者发现,在肿瘤中,负选择事件的数量极低。
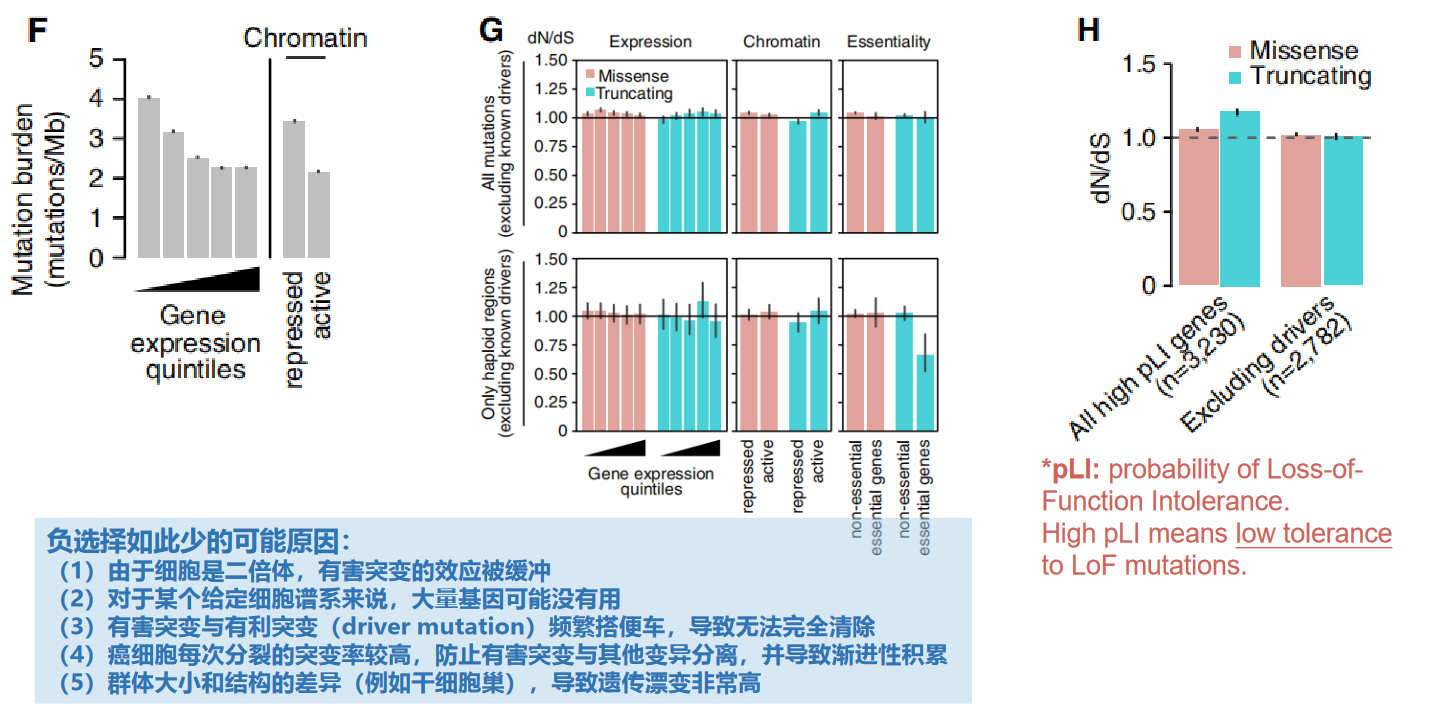
前面的统计聚焦于单个基因水平。作者想知道在基因集合的水平上,是否存在负选择的信号。这里作者将基因按照表达量或者染色质开放性做了分类,并做了统计。
- F: 基因表达量↑,突变负荷↓ ;染色质开放性↑,突变负荷↓,如Fig3F。(突变负荷=单位长度染色体携带的突变数量)
- G: 有人提出突变负荷↓是负选择的标志,但作者不认可, 因为不管基因表达水平如何,或者染色质开放性如何,dN/dS都没有显著差异【也就是说,这些区域仅仅是突变率低,和负选择没有关系】,如Fig3G所示。
- 但是,但是!对于拷贝数为1的基因组区域的必要基因,truncating mut的dN/dS确实显著低于1(G图右下角),大约三分之一的此类变异通过负选择而丢失。
- H: 作者单独提取了high pLI基因做分析。理论上,high pLI基因对突变容忍度很低,应该受到负选择,dN/dS<1才对。但是分析结果显示dN/dS接近1但略大于1,与预期不符。这表明在cancer中确实缺乏负选择。
负选择如此少的可能原因:
- (1)由于细胞是二倍体,有害突变的效应被缓冲
- (2)对于某个给定细胞谱系来说,大量基因可能没有用
- (3)有害突变频繁与有利突变(driver mutation)搭便车,导致无法完全清除
- (4)癌细胞每次分裂的突变率较高,防止有害突变与其他变异分离,并导致渐进性积累(所谓的穆勒棘轮效应)
- (5)群体大小和结构的差异(例如stem cell niche,干细胞巢),导致非常高的遗传漂变
4. 每个肿瘤的发生需要多少驱动突变
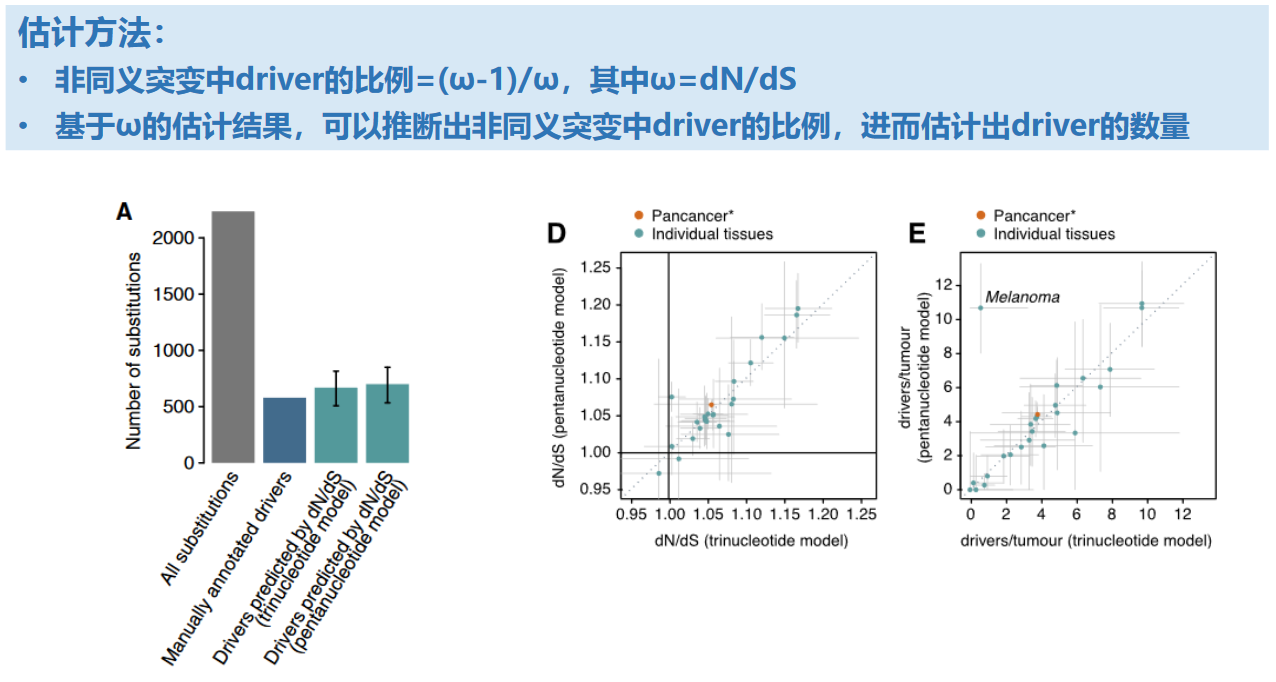
这一部分的工作想要回答的科学问题是“每个肿瘤的发生需要多少驱动突变?”
由于前面的工作已经证明肿瘤组织中的负选择很少,因此可以用dN/dS来估计驱动突变的数量。作者提出一种估计方法:
- 非同义突变中driver的比例=
(ω-1)/ω,其中ω=dN/dS - 基于ω的估计结果,可以推断出非同义突变中driver的比例,进而基于肿瘤的总突变数,从而估计出driver的数量
如A图(FigS4A),作者比较了在 breast cancer中使用上述方法的估计结果和manually annotation的实际结果,可以看出估计结果与annotation基本一致。
作者也讨论了考虑不同长度上下文的模型对估算结果的影响。FigS1D-E是作者另外用到的两个模型:trinucleotide模型(三核苷酸模型,考虑突变本身和上下游各1bp)和pentanucleotide模型(五核苷酸模型,考虑突变本身和上下游各2bp)。在绝大多数癌症中这两个模型差别都不显著,除了少数癌症如melanoma。
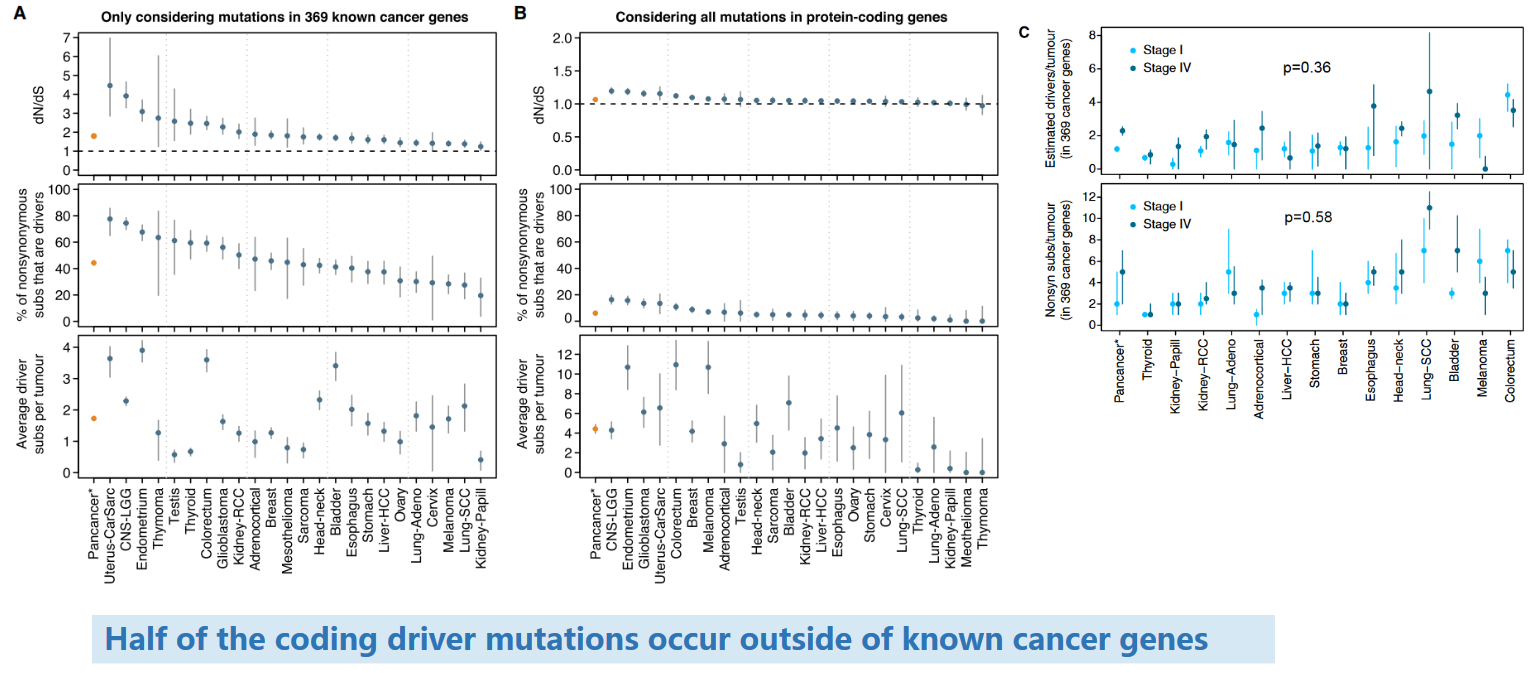
基于前面提出的这种估计方法,作者在不同肿瘤中估计了驱动突变的数量,结果如上图所示。
- A图是对已知cancer基因的估计结果。
- B图是在所有CDS区中的估计结果。可以看出来不同tumor的driver数量差别很大,但总体上平均每个肿瘤会携带4个左右的driver substitution。
- 这里的结果也表明,许多driver mutation其实并不在已知的cancer gene里面。还有许多未知等待挖掘。
- C图:为了评估在更晚期的癌症中,驱动突变的数量是否显著增加,作者对I期和IV期肿瘤进行了独立的估计。如图,其实早期肿瘤和晚期肿瘤的driver数量没有显著差异。
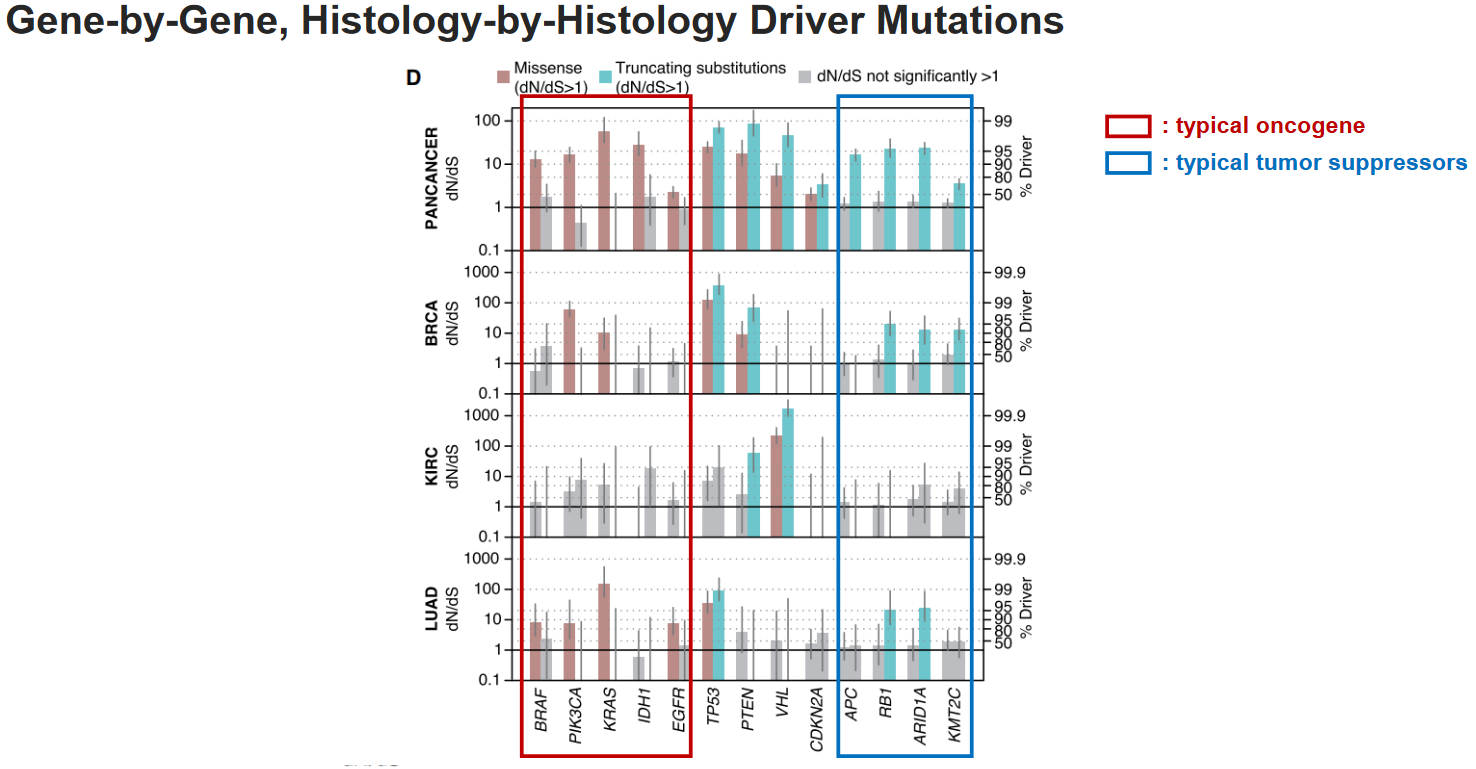
作者也单独挑出了一些比较著名的cancer gene,在不同肿瘤中对驱动突变的数量进行了估计,发现在肿瘤抑制基因中,错义替换是否可能是驱动突变的差异很大。
例如,上图中展示的几个原癌基因,其在肿瘤中的missense mutation多受到正选择( dN/dS>>1 );相比之下,几个典型的抑癌基因,missense mutation不受选择,更倾向于表现为乘客突变(即突变与否对tumor的发生没有太大作用,也因此不受选择压力),但truncating substitution会受到一些正选择。考虑到missense mutation通常会为蛋白质引入新的功能,而truncating substitution通常会导致蛋白质表达失败,肿瘤中驱动突变的这种分布模式就可以被我们所理解了。
5. 超突变肿瘤中的驱动突变是否更多?
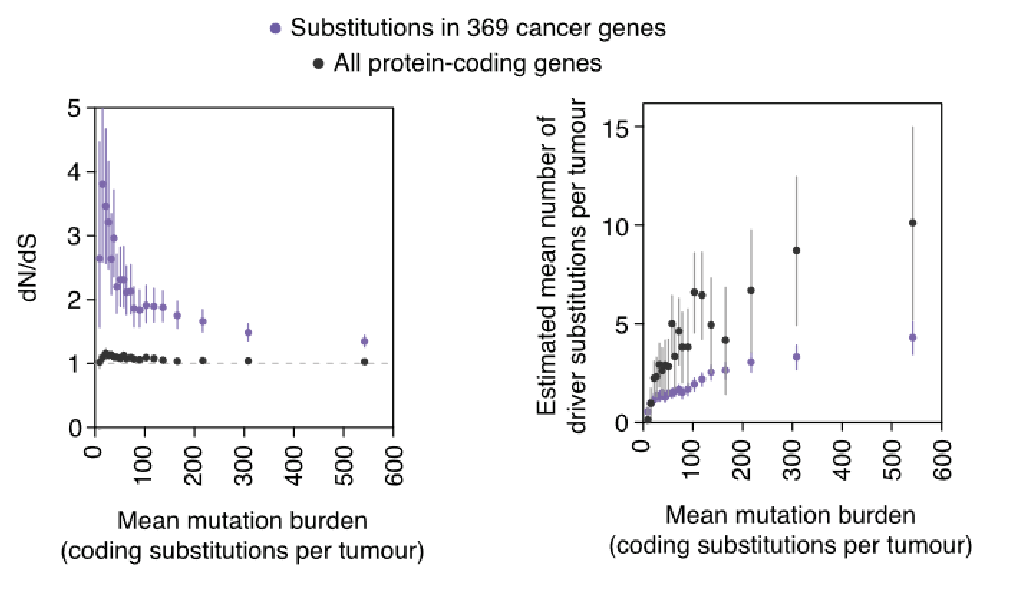
前面研究的是正常突变数的肿瘤,即平均每个肿瘤的coding区mutation少于500个(平均<17 mut/Mb),但少数肿瘤的突变数量远高于此,它们受到的选择压力如何仍然未知。这是这一小节想要研究的问题。
此外,还有一个科学问题值得回答:是否 超突变 导致了更高的driver mutation数量?亦或者 超突变 仅仅允许一个克隆比竞争克隆更快地获得固定的驱动补充?(说人话,就是在超突变肿瘤里面,对driver的需求是否也变得更高?)
上图统计了平均mut burden和dN/dS的关系。mut burden越高,dN/dS越趋近于1,但总体上依然高于1.。这导致随着突变负担的增加,驱动突变的数量也在增加,但二者的关系并非线性,增长率随着驱动突变的增加而在不断下降。
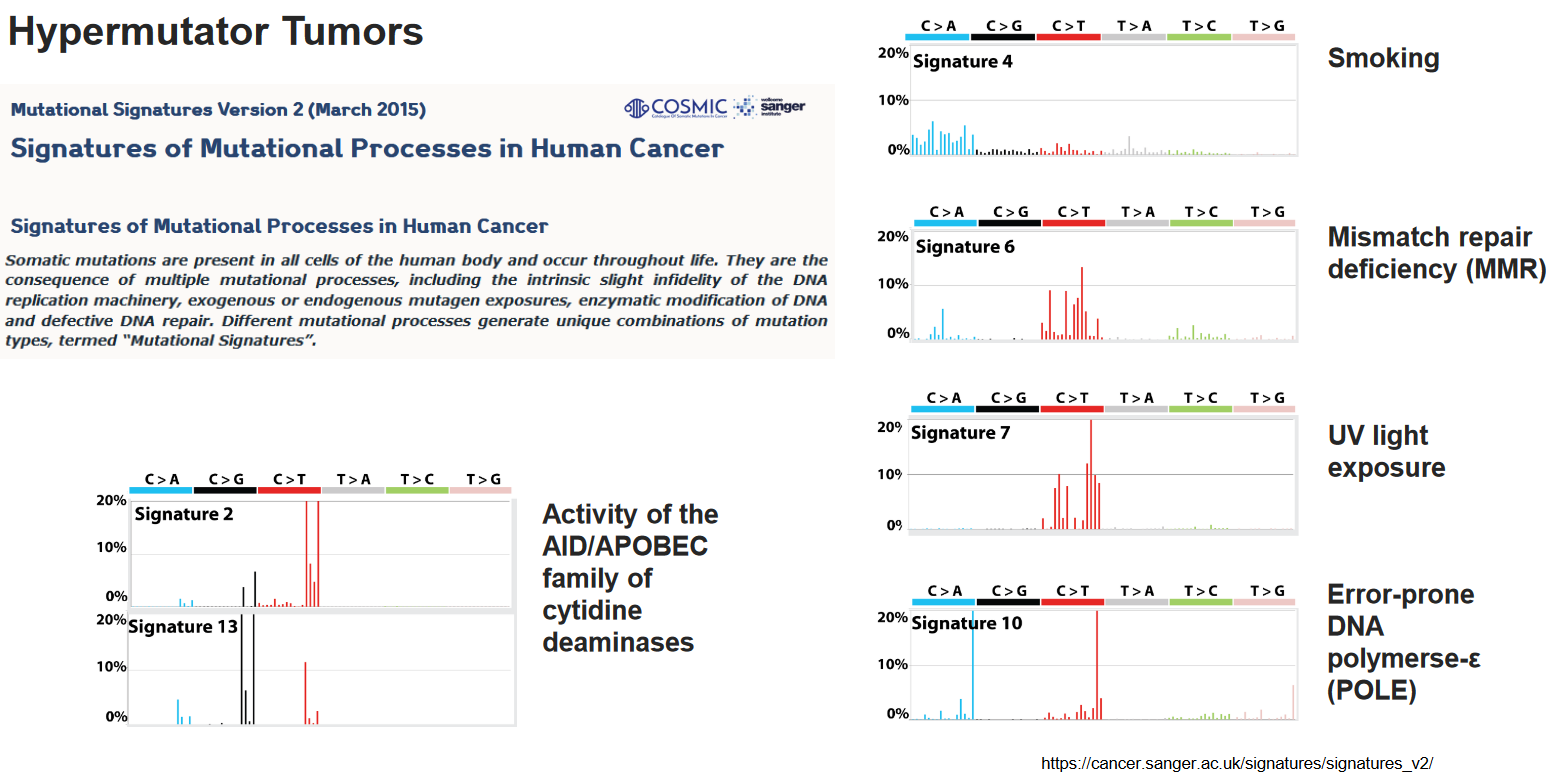
为了对这些突变做一些更精细的探究,作者基于COSMIC数据库的mutation signature 将这些hypermut肿瘤分为了图中所示的5个类别。如上图所示,COSMIC的signature描述的是一些致癌因素导致的突变过程,一种致癌因素会倾向于导致某些类型的突变更多的出现
- COSMIC数据库中的mutation signatures对应的是特定的突变过程,而不是单一的突变或肿瘤。每个突变特征通过96种碱基替换的比率来展示,可以反映不同的突变机制。一个肿瘤样本可能显示多个突变特征的组合,反映其复杂的突变背景和驱动机制。
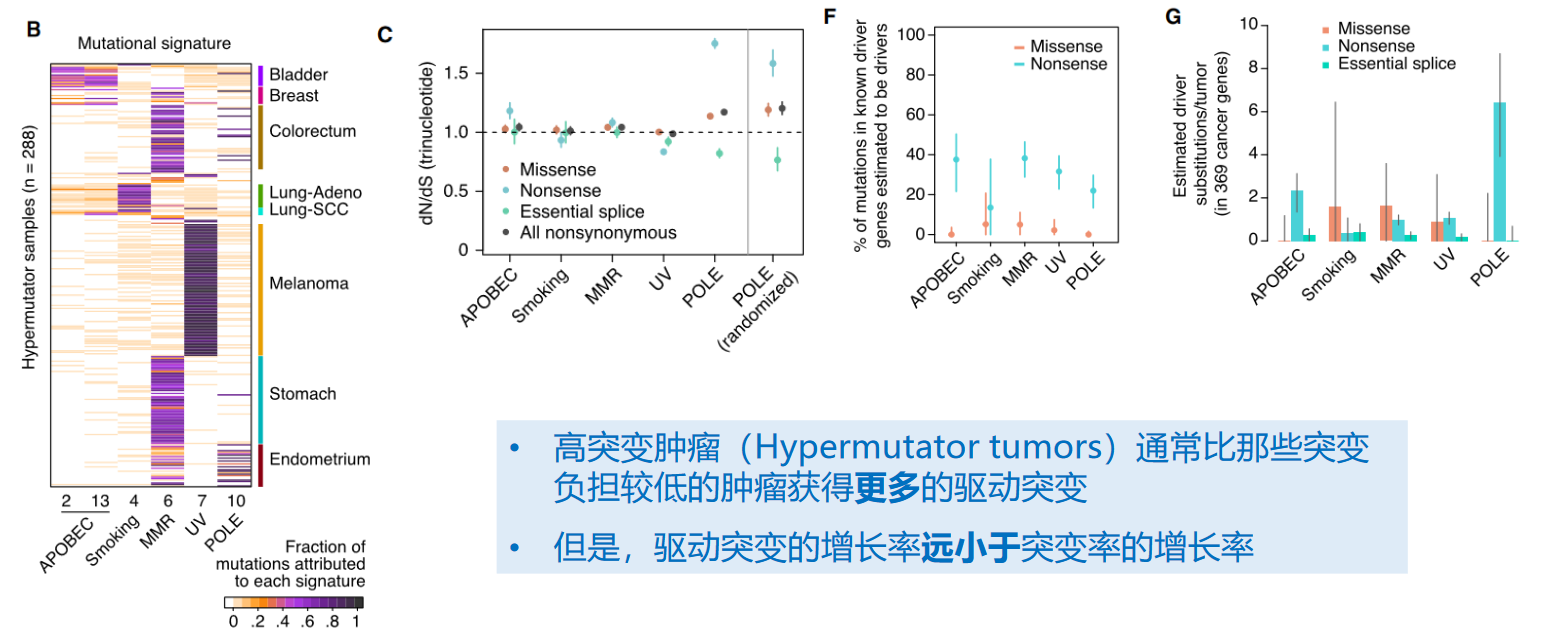
- B:基于COSMIC mutation signature(https://cancer.sanger.ac.uk/signatures/signatures_v2/) 的分类方法,按照dominant mutation process对突变进行划分。图中展示了各个肿瘤的mutation中不同signature的mutation的比例。
- C:分别提取了这些signature的tumor【感觉虽然B图是计算了每个tumor各个signature mut的占比,但到了C图,作者直接按B图的占比最高的signature,对tumor做划分了,后面的比较也都是在tumor层面做的分析】,并计算了dN/dS。属于大部分signature的mut实际上dN/dS依然在1附近,除了POLE这一个类型(DNA-pol-e的校对活性突变)。【UV这一类其实也有bias,但用pentanuclide model可以消除bias】。
- F:上述五种超突变肿瘤类型各自的driver mutation占比(注意分母为known driver gene,不是 all gene。因此虽然这个比例应该还是通过
ω-1/ω这个公式得到的,但是和Fig5C关系不大了)。 - G:上述五种超突变肿瘤类型各自的driver mutation数量。
- F和G放在一起看:作者发现,即使在这些hypermutation tumor(即突变率很高的肿瘤)中,能称之为driver mutation的突变依然很少。猜测这里的逻辑是,因为这些tumor中的dN/dS基本都为1,以至于在实际的missense mutation中driver mutation的占比很低。
结论:高突变肿瘤通常比那些突变负担较低的肿瘤获得更多的驱动突变,尽管其增加的比例远小于突变率的增加。
本文结论
作者开发了一个dN/dS的估计框架(dNdScv),这一框架基于泊松分布统计模型进行最大似然法估计,并使用广义线性模型对单个基因的背景突变率进行矫正,从而达到了很高的精确性。
作者将这一模型应用在了tumor tissue的选择压力分析当中。作者发现:
- 与germline不同,somatic的进化受到正选择的压力
- 接近99%的coding区突变对负选择有耐受性、可以逃脱负选择
- 开展了对各个肿瘤的全外显子组的对driver coding mutation数量的估计
- 半数coding driver mutation发生在已知cancer基因之外
扩展
需要注意的是,dN/dS只能检测编码区的突变,这也是本文的不足之处。2022年哈佛医学院的Eliezer M. Van Allen团队开发了一种方法(如下图),可以在非编码区检测somatic mutation,从而完善了肿瘤的突变检测方法。
所有参考文献均在正文的文本或图片中给出了链接,文末不再单独列出参考文献。